- Bleeding disordes in children

Содержание
- 2. The bleeding disorders is a group of diseases with increased bleeding, which is based on disorders
- 3. Overview of Haemostasis INJURY Collagen Exposure Platelet Adhesion and release reaction Platelet aggregation VASOCONSTRICTION Serotonin Platelet
- 7. This disorder may be due to: A functional deficiency in the procoagulant mechanism. This may involve:
- 8. Three types can be broadly identified: “Coagulation-defect bleeds” “Purpuric-type bleeds” Mixed bleeds.
- 9. Differential diagnosis between coagulation disorders and purpuric disorders
- 13. Bleeding history The bleeding history forms the basis of the laboratory tests and therapy. Asking patients
- 14. Physical Examination Several clinical as well as laboratory features help differentiate clinical disorders associated with qualitative
- 15. The usual lesions manifest spontaneous petechiae and ecchymoses, which result from a breakdown of the anatomic
- 16. Gastrointestinal and genitourinary bleeding may occur spontaneously with abnormalities of platelets and/or coagulation factors. Deep hematomas,
- 17. LABORATORY EVALUATION OF HEMOSTATIC DISORDERS Understanding of the physiology of primary and secondary hemostasis is important
- 18. The platelet count is performed to detect thrombocytopenia, which is defined as a platelet count of
- 19. The bleeding time is defined as the time between the infliction of a small standard cut
- 20. The various methods for performing the bleeding time are basically modifications of two techniques: the bleeding
- 21. The prothrombin time (PT) The prothrombin time may be prolonged because of a deficiency of a
- 22. The aPTT (activated partial thromboplastin time) In the old “cascade” theory of coagulation, the aPTT involves
- 23. The thrombin time (TT) as part of the screening procedures. The thrombin time will be prolonged
- 24. Interpretation of the Screening Tests of Hemostasis Discrimination of the majority of the inherited and acquired
- 25. A prolonged PT and a normal aPTT and TT may indicate a factor VII deficiency. Inherited
- 26. Most congenital deficiencies are single, whereas acquired abnormalities caused by vitamin K deficiency, liver disease, disseminated
- 27. PLATELET DISORDERS Though platelets are classified as cells, they are actually cytoplasmatic fragments derived from megakaryocytes
- 28. Thrombocytopenia thrombocytopenia occurs when the platelet amount drops below 150,000/μL. The bleeding usually occurs when the
- 29. Classification of Thrombocytopenia
- 30. MDS, myelodysplastic syndrome; SLE, systemic lupus erythematosus; CLL, chronic lymphocytic leukemia; DIC, disseminated intravascular coagulation.
- 31. Thrombocytopenia Due to Decreased Platelet Production Thrombocytopenia due to decreased platelet production means that the bone
- 33. Thrombocytopenia Due to Increased Platelet Destruction Isolated thrombocytopenia is caused by increased platelet destruction. In these
- 34. Immune Thrombocytopenia Immune thrombocytopenia is an increase of platelet destruction caused by immunological mechanisms. The sensitization
- 35. Idiopathic Thrombocytopenic Purpura Acute ITP in children is equally common in boys and girls, has its
- 36. SLE 5% APS 2% CVID 1% CLL 2% Evan’s 2% ALPS, post-tx 1% HIV 1% Hep
- 37. Causes The exact causes of ITP are yet unknown, but there is currently research going on
- 38. Theories Three most common theories for ITP are: The Microbial Trigger Theory The Molecular Mimicry Theory
- 39. The Microbial Trigger Theory Related to the destruction of platelets to a chemical called interleuken 12
- 40. The Molecular Mimicry Theory This theory says that someone can develop ITP when the bodies T-helper
- 41. Free Radical Damage Theory DNA is damaged by “free radicals”. Free radicals are compounds that build
- 42. Pathophysiology ITP is caused by an autoantibody - in generally IgG - that binds to specific
- 43. Increased platelet destruction & Inadequate platelet production Two Processes Involved in Immune Thromcytopenic Purpura Platelets Antibody
- 44. Classification of ITP Primary ITP: Idiopathic - etiology is unknown; No clinically evident secondary form. Secondary
- 45. Classification of ITP Secondary ITP Antiphospholipid syndrome; Autoimmune thrombocytopenia (e.g., Evans syndrome); Common variable immune deficiency;
- 46. Newly diagnosed (acute) ITP: Less than 6 months; Chronic ITP: More than 6-12 months; Refractory ITP:
- 47. Symptoms of ITP Excessive bleeding after minor injuries; Spontaneous bleeding from the mouth and nose; Unexplainable
- 48. Depression and ITP ITP is accompanied by short term or more permanent depression. This is because
- 49. Diagnosis of ITP Platelet count: Less than 100 x 109/L (rather than 150 x 109/L) Medical
- 50. Indications for Treatment American Society of Hematology (ASH) suggests: Platelet amount > 30 x 109/L usually
- 51. Goals of Treatment Obtain a hemostatic platelet amount to prevent bleeding: Individualized to the patient; Minimizing
- 52. ITP Treatment Options 1st line: Corticosteroids; Intravenous immunoglobulins (IVIG); 2nd Line: Splenecotomy; Thrombopoietin Receptor Agonists (in
- 53. ITP Treatment Options: Corticosteroids Prednisolone: Mechanism of Action: Impair clearance of platelets in the bone marrow
- 54. ITP Treatment Options: IVIG IVIG: Mechanism of action: Undefined and potentially multifactorial Dose: Variable regimen Dose:
- 55. ITP Treatment Options: Splenectomy Splenectomy: Mechanism of action: Removes a primary site of platelet destruction and
- 56. EMERGENCY TREATMENT OF ITP Platelet transfusion + high dose steroids Platelet transfusion + continuous IVIG Antifibrinolytics
- 57. Neonatal Purpura Neonatal thrombocytopenia may develop due to isoimmunization of the mother against fetal platelets with
- 58. INHERITED DISORDERS OF COAGULATION There is a large number of inherited disorders of coagulation; however, only
- 59. von Willebrand’s Disease (vWD) von Willebrand disease is the most common inherited disorder of primary hemostasis.
- 60. Three main subtypes of vWD have been defined: Type 1: the most common (≥70% of cases
- 61. Type 2: In Type 2 vWD, there is a qualitative defect in vWF. Several different subtypes
- 62. Type 3: In Type 3 vWD, there is a total or near-total absence of vWF in
- 63. Clinical Manifestations of von Willebrand’s Disease Most cases of vWD present with the typical picture of
- 64. The laboratory manifestations of vWD can also be highly variable; sometimes laboratory tests must be repeated
- 65. Laboratory Diagnosis of von Willebrand’s Disease The most important diagnostic tests for vWD are the bleeding
- 66. In vWD Type 1, the BT will usually be prolonged. The PTT may also be slightly
- 67. Type 2A is diagnosed by demonstrating an absence of the high-molecular-weight multimers by agarose gel electrophoresis.
- 68. Diagnosis of Type 2M requires sophisticated techniques, which are not widely available; therefore, specimens must usually
- 69. Treatment of von Willebrand’s Disease Most cases of vWD Type 1 can be very successfully treated
- 71. Indications for clotting factor concentrate administration in vWD Type 2 or 3 vWD: Active bleeding; Surgery
- 73. The Hemophilias The hemophilias are inherited disorders of the coagulation cascade. Deficiency of factor VIII (hemophilia
- 74. Hemophilia A and B are both inherited as X-linked recessive: women are carriers, men develop the
- 75. Clinical Features The clinical features of hemophilia A and B are identical. The manifestations are those
- 76. Muscle hematoma (pseudotumor) Hemarthrosis (joint bleeding)
- 77. LONG-TERM COMPLICATIONS OF HEMOPHILIA
- 78. Hemophilic arthropathy “Target joint” = irreversibly damaged joint with vicious cycle of injury and repeated bleeding
- 79. Intracranial bleeding is anespecially serious complication, and even minor head trauma in a severe hemophiliac should
- 80. Children with severe hemophilia usually begin to have problems at about 9 to 12 months of
- 81. Most patients will have a family history of pathologic bleeding on the maternal side, including maternal
- 82. Hemophilia: Factor Level versus Severity* *Generally applies to both factor VIII and factor IX deficiencies; may
- 83. Laboratory Diagnosis The PTT is prolonged; the PT is normal. Mixing studies show correction of the
- 84. Treatment Treatment depends on which factor is deficient, the severity of the deficiency, and the nature
- 85. Always be sure to determine which factor the patient is deficient in before you start replacement
- 86. Dosing clotting factor concentrate 1 U/kg of factor VIII should increase plasma level by about 2%
- 87. Give factor q 12 hours for 2-3 days after major surgery, continue with daily infusions for
- 88. Liver disease in hemophilia Hepatitis C is still a problem, though incidence is falling with safer
- 89. ACQUIRED FACTOR VIII DEFICIENCY Due to antibody to factor VIII (most common autoimmune factor deficiency) Most
- 91. Скачать презентацию

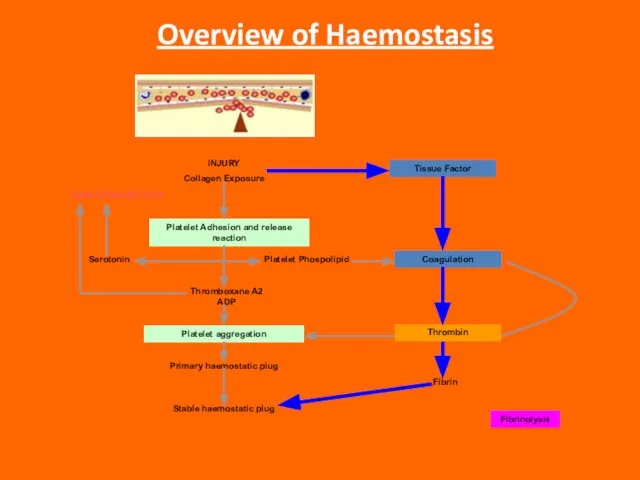
Слайд 3Overview of Haemostasis
INJURY
Collagen Exposure
Platelet Adhesion and release reaction
Platelet aggregation
VASOCONSTRICTION
Serotonin
Platelet Phospolipid
Thromboxane A2 ADP
Primary
Overview of Haemostasis
INJURY
Collagen Exposure
Platelet Adhesion and release reaction
Platelet aggregation
VASOCONSTRICTION
Serotonin
Platelet Phospolipid
Thromboxane A2 ADP
Primary
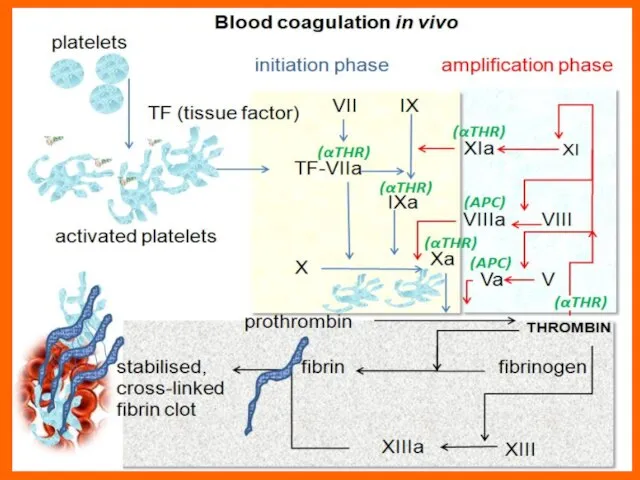
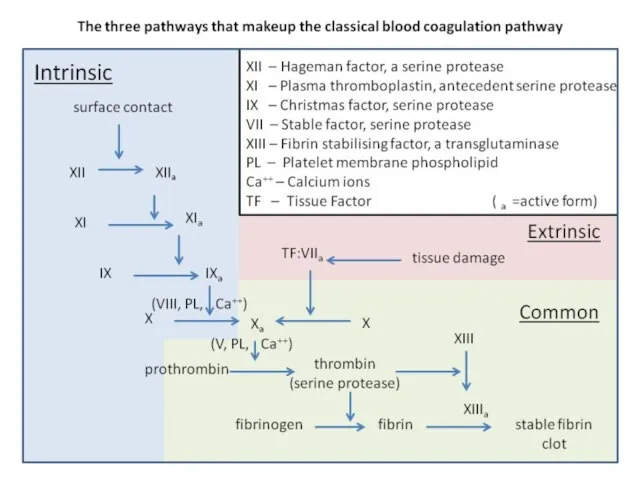
Слайд 7This disorder may be due to:
A functional deficiency in the procoagulant mechanism.
This disorder may be due to:
A functional deficiency in the procoagulant mechanism.

Слайд 8Three types can be broadly identified:
“Coagulation-defect bleeds”
“Purpuric-type bleeds”
Mixed bleeds.
Three types can be broadly identified:
“Coagulation-defect bleeds”
“Purpuric-type bleeds”
Mixed bleeds.

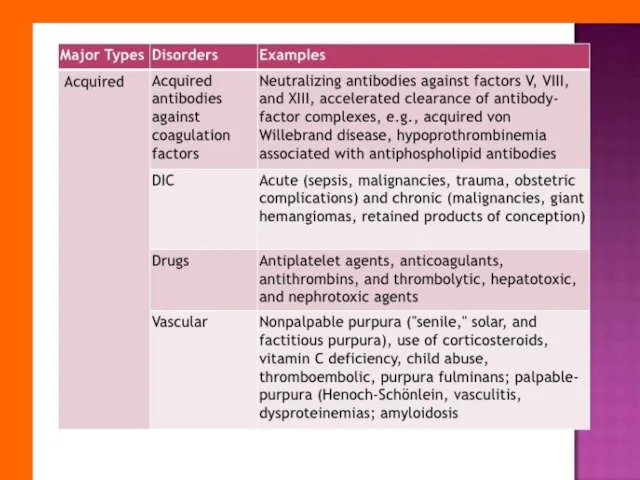
Слайд 9Differential diagnosis between coagulation disorders
and purpuric disorders
Differential diagnosis between coagulation disorders
and purpuric disorders



Слайд 13Bleeding history
The bleeding history forms the basis of the laboratory tests and
Bleeding history
The bleeding history forms the basis of the laboratory tests and

Слайд 14Physical Examination
Several clinical as well as laboratory features help differentiate clinical disorders
Physical Examination
Several clinical as well as laboratory features help differentiate clinical disorders

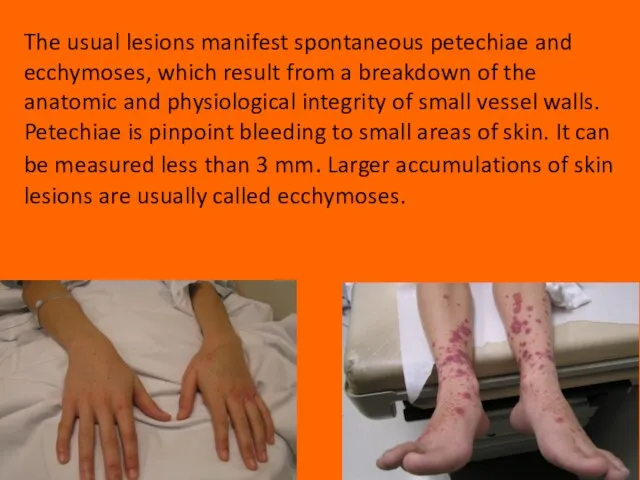
Слайд 15The usual lesions manifest spontaneous petechiae and ecchymoses, which result from a
The usual lesions manifest spontaneous petechiae and ecchymoses, which result from a
Слайд 16Gastrointestinal and genitourinary bleeding may occur spontaneously with abnormalities of platelets and/or
Gastrointestinal and genitourinary bleeding may occur spontaneously with abnormalities of platelets and/or

Слайд 17LABORATORY EVALUATION OF HEMOSTATIC DISORDERS
Understanding of the physiology of primary and secondary
LABORATORY EVALUATION OF HEMOSTATIC DISORDERS
Understanding of the physiology of primary and secondary

Слайд 18The platelet count
is performed to detect thrombocytopenia, which is defined as a
The platelet count
is performed to detect thrombocytopenia, which is defined as a

Слайд 19The bleeding time
is defined as the time between the infliction of a
The bleeding time
is defined as the time between the infliction of a

Слайд 20The various methods for performing the bleeding time are basically modifications of
The various methods for performing the bleeding time are basically modifications of

Слайд 21The prothrombin time (PT)
The prothrombin time may be prolonged because of a
The prothrombin time (PT)
The prothrombin time may be prolonged because of a

Слайд 22The aPTT (activated partial thromboplastin time)
In the old “cascade” theory of coagulation,
The aPTT (activated partial thromboplastin time)
In the old “cascade” theory of coagulation,

Слайд 23The thrombin time (TT)
as part of the screening procedures. The thrombin time
The thrombin time (TT)
as part of the screening procedures. The thrombin time

Слайд 24Interpretation of the Screening Tests of Hemostasis
Discrimination of the majority of the
Interpretation of the Screening Tests of Hemostasis
Discrimination of the majority of the

Слайд 25 A prolonged PT and a normal aPTT and TT may
indicate
A prolonged PT and a normal aPTT and TT may
indicate

Слайд 26Most congenital deficiencies are single, whereas acquired abnormalities caused by vitamin K
Most congenital deficiencies are single, whereas acquired abnormalities caused by vitamin K

Слайд 27PLATELET DISORDERS
Though platelets are classified as cells, they are actually cytoplasmatic fragments
PLATELET DISORDERS
Though platelets are classified as cells, they are actually cytoplasmatic fragments

Слайд 28Thrombocytopenia
thrombocytopenia occurs when the platelet amount drops below 150,000/μL. The bleeding usually
Thrombocytopenia
thrombocytopenia occurs when the platelet amount drops below 150,000/μL. The bleeding usually

Слайд 29Classification of Thrombocytopenia
Classification of Thrombocytopenia

Слайд 30MDS, myelodysplastic syndrome; SLE, systemic lupus erythematosus; CLL, chronic
lymphocytic leukemia; DIC, disseminated
MDS, myelodysplastic syndrome; SLE, systemic lupus erythematosus; CLL, chronic
lymphocytic leukemia; DIC, disseminated

Слайд 31Thrombocytopenia Due to Decreased Platelet Production
Thrombocytopenia due to decreased platelet production means
Thrombocytopenia Due to Decreased Platelet Production
Thrombocytopenia due to decreased platelet production means


Слайд 33Thrombocytopenia Due to Increased Platelet Destruction
Isolated thrombocytopenia is caused by increased platelet
Thrombocytopenia Due to Increased Platelet Destruction
Isolated thrombocytopenia is caused by increased platelet

Слайд 34Immune Thrombocytopenia
Immune thrombocytopenia is an increase of platelet destruction caused by immunological
Immune Thrombocytopenia
Immune thrombocytopenia is an increase of platelet destruction caused by immunological

Слайд 35Idiopathic Thrombocytopenic Purpura
Acute ITP in children is equally common in boys and
Idiopathic Thrombocytopenic Purpura
Acute ITP in children is equally common in boys and

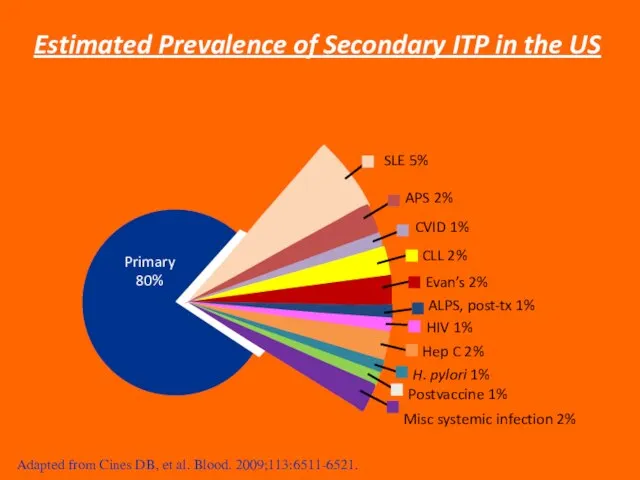
Слайд 36SLE 5%
APS 2%
CVID 1%
CLL 2%
Evan’s 2%
ALPS, post-tx 1%
HIV 1%
Hep C 2%
H. pylori
SLE 5%
APS 2%
CVID 1%
CLL 2%
Evan’s 2%
ALPS, post-tx 1%
HIV 1%
Hep C 2%
H. pylori
Слайд 37Causes
The exact causes of ITP are yet unknown, but there is currently
Causes
The exact causes of ITP are yet unknown, but there is currently

Слайд 38Theories
Three most common theories for ITP are:
The Microbial Trigger Theory
The Molecular Mimicry
Theories
Three most common theories for ITP are:
The Microbial Trigger Theory
The Molecular Mimicry

Слайд 39The Microbial Trigger Theory
Related to the destruction of platelets to a chemical
The Microbial Trigger Theory
Related to the destruction of platelets to a chemical

Слайд 40The Molecular Mimicry Theory
This theory says that someone can develop ITP when
The Molecular Mimicry Theory
This theory says that someone can develop ITP when

Слайд 41Free Radical Damage Theory
DNA is damaged by “free radicals”.
Free radicals are compounds
Free Radical Damage Theory
DNA is damaged by “free radicals”.
Free radicals are compounds

Слайд 42Pathophysiology
ITP is caused by an autoantibody - in generally IgG - that
Pathophysiology
ITP is caused by an autoantibody - in generally IgG - that

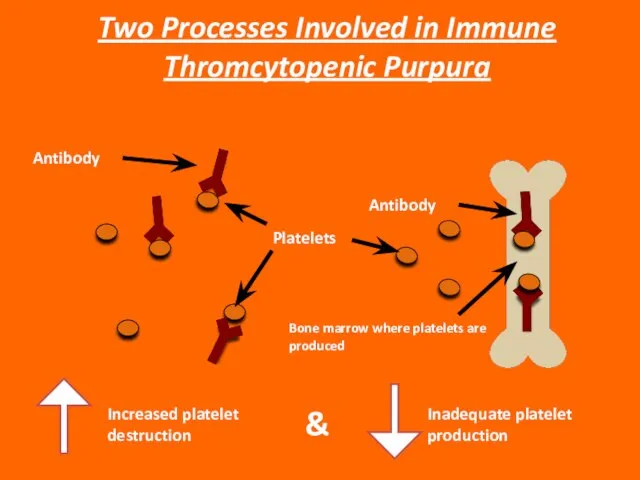
Слайд 43Increased platelet destruction
&
Inadequate platelet production
Two Processes Involved in Immune Thromcytopenic Purpura
Platelets
Antibody
Antibody
Bone marrow
Increased platelet destruction
&
Inadequate platelet production
Two Processes Involved in Immune Thromcytopenic Purpura
Platelets
Antibody
Antibody
Bone marrow
Слайд 44Classification of ITP
Primary ITP:
Idiopathic - etiology is unknown;
No clinically evident secondary
Classification of ITP
Primary ITP:
Idiopathic - etiology is unknown;
No clinically evident secondary

Слайд 45Classification of ITP
Secondary ITP
Antiphospholipid syndrome;
Autoimmune thrombocytopenia (e.g., Evans syndrome);
Common variable immune
Classification of ITP
Secondary ITP
Antiphospholipid syndrome;
Autoimmune thrombocytopenia (e.g., Evans syndrome);
Common variable immune

Слайд 46Newly diagnosed (acute) ITP:
Less than 6 months;
Chronic ITP:
More than 6-12 months;
Refractory ITP:
Treatment
Newly diagnosed (acute) ITP:
Less than 6 months;
Chronic ITP:
More than 6-12 months;
Refractory ITP:
Treatment

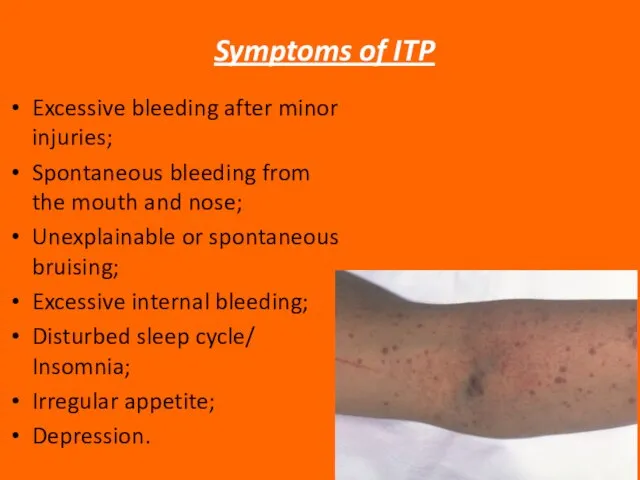
Слайд 47 Symptoms of ITP
Excessive bleeding after minor injuries;
Spontaneous bleeding from the
Symptoms of ITP
Excessive bleeding after minor injuries;
Spontaneous bleeding from the
Слайд 48Depression and ITP
ITP is accompanied by short term or more permanent depression.
Depression and ITP
ITP is accompanied by short term or more permanent depression.

Слайд 49Diagnosis of ITP
Platelet count:
Less than 100 x 109/L (rather than 150 x
Diagnosis of ITP
Platelet count:
Less than 100 x 109/L (rather than 150 x

Слайд 50Indications for Treatment
American Society of Hematology (ASH) suggests:
Platelet amount > 30 x
Indications for Treatment
American Society of Hematology (ASH) suggests:
Platelet amount > 30 x

Слайд 51Goals of Treatment
Obtain a hemostatic platelet amount to
prevent bleeding:
Individualized to
Goals of Treatment
Obtain a hemostatic platelet amount to
prevent bleeding:
Individualized to

Слайд 52ITP Treatment Options
1st line:
Corticosteroids;
Intravenous immunoglobulins (IVIG);
2nd Line:
Splenecotomy;
Thrombopoietin Receptor Agonists (in adults);
Rituximab (in
ITP Treatment Options
1st line:
Corticosteroids;
Intravenous immunoglobulins (IVIG);
2nd Line:
Splenecotomy;
Thrombopoietin Receptor Agonists (in adults);
Rituximab (in

Слайд 53ITP Treatment Options:
Corticosteroids
Prednisolone:
Mechanism of Action:
Impair clearance of platelets in the bone
ITP Treatment Options:
Corticosteroids
Prednisolone:
Mechanism of Action:
Impair clearance of platelets in the bone

Слайд 54ITP Treatment Options:
IVIG
IVIG:
Mechanism of action:
Undefined and potentially multifactorial
Dose:
Variable regimen
Dose:
ITP Treatment Options:
IVIG
IVIG:
Mechanism of action:
Undefined and potentially multifactorial
Dose:
Variable regimen
Dose:

Слайд 55ITP Treatment Options:
Splenectomy
Splenectomy:
Mechanism of action:
Removes a primary site of platelet destruction and
ITP Treatment Options:
Splenectomy
Splenectomy:
Mechanism of action:
Removes a primary site of platelet destruction and

Слайд 56EMERGENCY TREATMENT OF ITP
Platelet transfusion + high dose steroids
Platelet transfusion + continuous
EMERGENCY TREATMENT OF ITP
Platelet transfusion + high dose steroids
Platelet transfusion + continuous

Слайд 57Neonatal Purpura
Neonatal thrombocytopenia may develop due to isoimmunization of the mother against
Neonatal Purpura
Neonatal thrombocytopenia may develop due to isoimmunization of the mother against

Слайд 58INHERITED DISORDERS OF COAGULATION
There is a large number of inherited disorders of
INHERITED DISORDERS OF COAGULATION
There is a large number of inherited disorders of

Слайд 59von Willebrand’s Disease (vWD)
von Willebrand disease is the most common inherited disorder
von Willebrand’s Disease (vWD)
von Willebrand disease is the most common inherited disorder

Слайд 60Three main subtypes of vWD have been defined:
Type 1: the most common
Three main subtypes of vWD have been defined:
Type 1: the most common

Слайд 61Type 2: In Type 2 vWD, there is a qualitative defect in
Type 2: In Type 2 vWD, there is a qualitative defect in

Слайд 62Type 3: In Type 3 vWD, there is a total or near-total
Type 3: In Type 3 vWD, there is a total or near-total

Слайд 63Clinical Manifestations of von Willebrand’s Disease
Most cases of vWD present with the
Clinical Manifestations of von Willebrand’s Disease
Most cases of vWD present with the

Слайд 64The laboratory manifestations of vWD can also be highly variable; sometimes laboratory
The laboratory manifestations of vWD can also be highly variable; sometimes laboratory

Слайд 65Laboratory Diagnosis of von Willebrand’s Disease
The most important diagnostic tests for vWD
Laboratory Diagnosis of von Willebrand’s Disease
The most important diagnostic tests for vWD

Слайд 66In vWD Type 1, the BT will usually be prolonged. The PTT
In vWD Type 1, the BT will usually be prolonged. The PTT

Слайд 67Type 2A is diagnosed by demonstrating an absence of the high-molecular-weight multimers
Type 2A is diagnosed by demonstrating an absence of the high-molecular-weight multimers

Слайд 68Diagnosis of Type 2M requires sophisticated techniques, which are not widely available;
Diagnosis of Type 2M requires sophisticated techniques, which are not widely available;

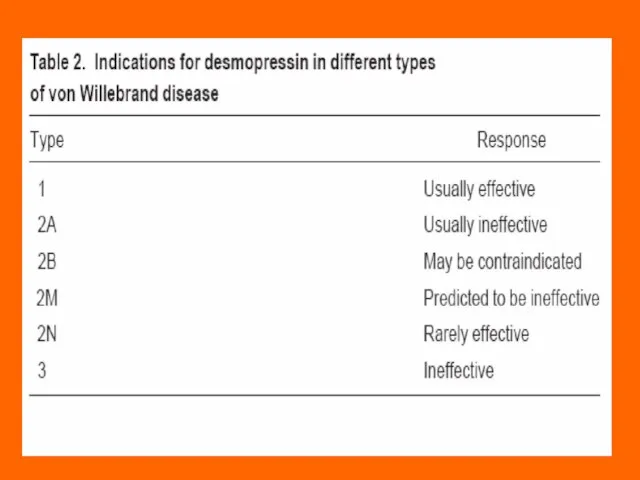
Слайд 69Treatment of von Willebrand’s Disease
Most cases of vWD Type 1 can be
Treatment of von Willebrand’s Disease
Most cases of vWD Type 1 can be

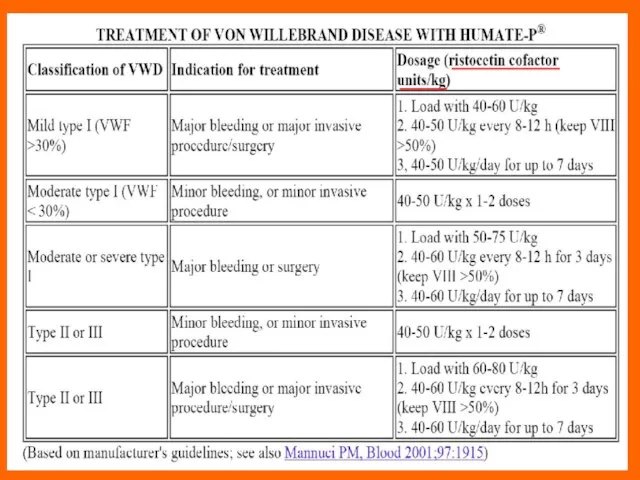
Слайд 71Indications for clotting factor concentrate administration in vWD
Type 2 or 3 vWD:
Active
Indications for clotting factor concentrate administration in vWD
Type 2 or 3 vWD:
Active

Слайд 73The Hemophilias
The hemophilias are inherited disorders of the coagulation cascade. Deficiency of
The Hemophilias
The hemophilias are inherited disorders of the coagulation cascade. Deficiency of

Слайд 74Hemophilia A and B are both inherited as X-linked recessive: women are
Hemophilia A and B are both inherited as X-linked recessive: women are

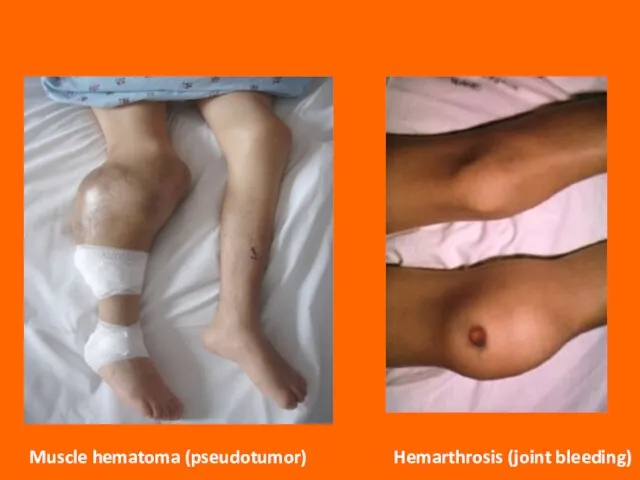
Слайд 75Clinical Features
The clinical features of hemophilia A and B are identical. The
Clinical Features
The clinical features of hemophilia A and B are identical. The

Слайд 76Muscle hematoma (pseudotumor)
Hemarthrosis (joint bleeding)
Muscle hematoma (pseudotumor)
Hemarthrosis (joint bleeding)
Слайд 77LONG-TERM COMPLICATIONS OF HEMOPHILIA
LONG-TERM COMPLICATIONS OF HEMOPHILIA
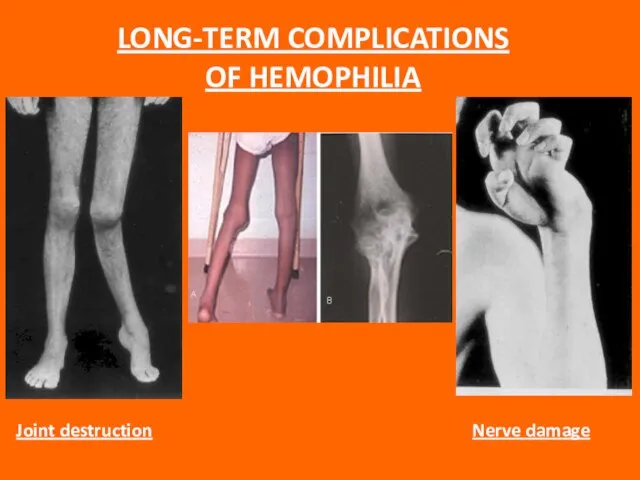
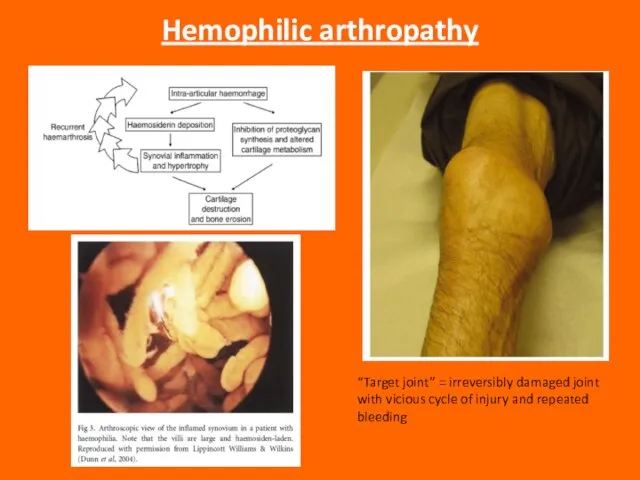
Слайд 78Hemophilic arthropathy
“Target joint” = irreversibly damaged joint with vicious cycle of injury
Hemophilic arthropathy
“Target joint” = irreversibly damaged joint with vicious cycle of injury
Слайд 79Intracranial bleeding is anespecially serious complication, and even minor head trauma in
Intracranial bleeding is anespecially serious complication, and even minor head trauma in

Слайд 80Children with severe hemophilia usually begin to have problems at about 9
Children with severe hemophilia usually begin to have problems at about 9

Слайд 81Most patients will have a family history of pathologic bleeding on the
Most patients will have a family history of pathologic bleeding on the

Слайд 82Hemophilia: Factor Level versus Severity*
*Generally applies to both factor VIII and factor
Hemophilia: Factor Level versus Severity*
*Generally applies to both factor VIII and factor

Слайд 83Laboratory Diagnosis
The PTT is prolonged; the PT is normal. Mixing studies show
Laboratory Diagnosis
The PTT is prolonged; the PT is normal. Mixing studies show

Слайд 84Treatment
Treatment depends on which factor is deficient, the severity of the deficiency,
Treatment
Treatment depends on which factor is deficient, the severity of the deficiency,

Слайд 85Always be sure to determine which factor the patient is deficient in
Always be sure to determine which factor the patient is deficient in

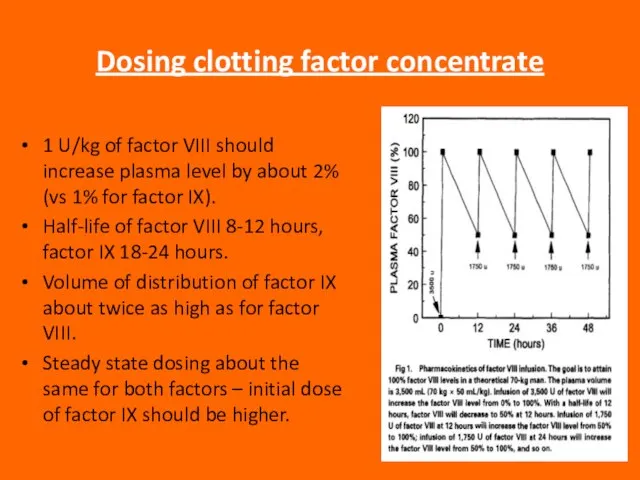
Слайд 86Dosing clotting factor concentrate
1 U/kg of factor VIII should increase plasma level
Dosing clotting factor concentrate
1 U/kg of factor VIII should increase plasma level
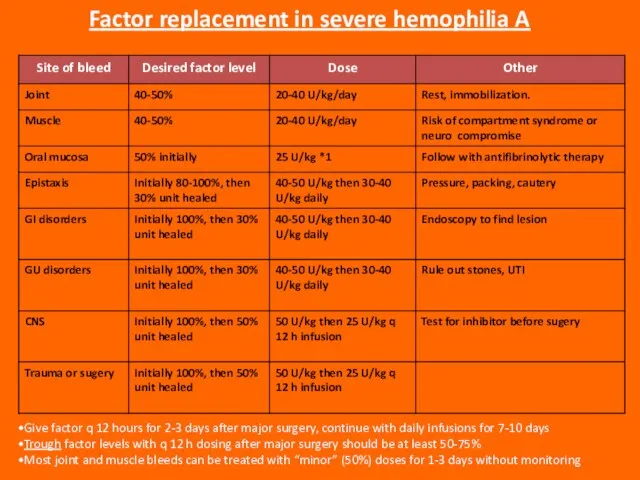
Слайд 87Give factor q 12 hours for 2-3 days after major surgery, continue
Give factor q 12 hours for 2-3 days after major surgery, continue
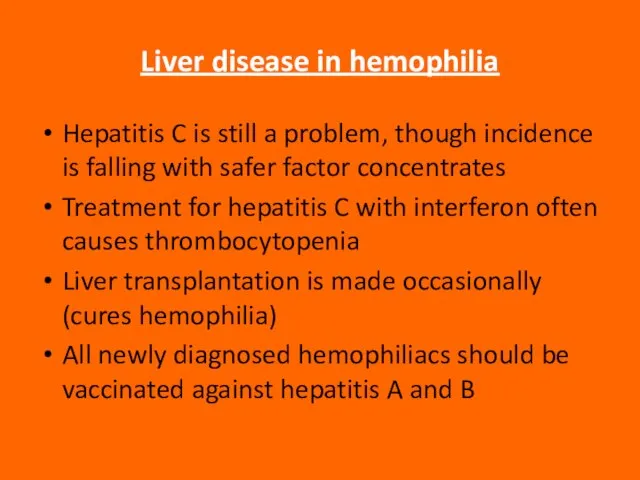
Слайд 88Liver disease in hemophilia
Hepatitis C is still a problem, though incidence is
Liver disease in hemophilia
Hepatitis C is still a problem, though incidence is
Слайд 89ACQUIRED FACTOR VIII DEFICIENCY
Due to antibody to factor VIII (most common autoimmune
ACQUIRED FACTOR VIII DEFICIENCY
Due to antibody to factor VIII (most common autoimmune

 Добро пожаловать в игру «Вверх по радуге»на этапе «В мире чисел»!!!
Добро пожаловать в игру «Вверх по радуге»на этапе «В мире чисел»!!! Туристическая компания «РОЗА ВЕТРОВ»
Туристическая компания «РОЗА ВЕТРОВ» Методическая работа направлена на всестороннее повышение квалификации и профессионального мастерства каждого профессионально-п
Методическая работа направлена на всестороннее повышение квалификации и профессионального мастерства каждого профессионально-п General anatomy of the опорно-impellent device
General anatomy of the опорно-impellent device Фальцеосадочная машина F 300-2 PLUS
Фальцеосадочная машина F 300-2 PLUS Эндокринные железы
Эндокринные железы Полиэтническая школа – пространство для диалога
Полиэтническая школа – пространство для диалога Директор МОУ СОШ №1 с.Кизляр Айдарова Р.У.
Директор МОУ СОШ №1 с.Кизляр Айдарова Р.У. Виды портрета человека
Виды портрета человека Суггестивные методы психокоррекции
Суггестивные методы психокоррекции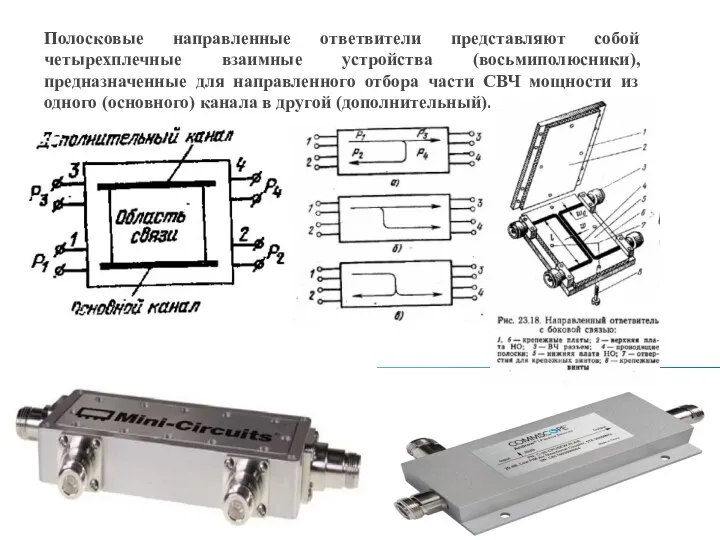 Направленные ответвители
Направленные ответвители Загадки Джоконды
Загадки Джоконды Наш двор
Наш двор Игра народов Коми Беляша
Игра народов Коми Беляша Годовой отчет учителя логопеда
Годовой отчет учителя логопеда Михаил Юрьевич Лермонтов «Герой нашего времени»
Михаил Юрьевич Лермонтов «Герой нашего времени» Управление персоналомTempus JEP - 27081 - 2006 «Поддержка и продвижение активного внедрения ИКТ в университетское управление в российских
Управление персоналомTempus JEP - 27081 - 2006 «Поддержка и продвижение активного внедрения ИКТ в университетское управление в российских  Презентация на тему Развитие жизни на планете
Презентация на тему Развитие жизни на планете Северная Аврора
Северная Аврора Интерактивное взаимодействие со зрителями Танец с огнем, веселая клоунада и немного мистики объединяются в сюжетную историю Наше
Интерактивное взаимодействие со зрителями Танец с огнем, веселая клоунада и немного мистики объединяются в сюжетную историю Наше  Использование ИКТ на уроках русского языка , литературы и во внеклассной деятельности.
Использование ИКТ на уроках русского языка , литературы и во внеклассной деятельности. «БОРОДИНО»
«БОРОДИНО» Виды услуг, оказываемых исследовательскими агентствами
Виды услуг, оказываемых исследовательскими агентствами Верхняя и нижняя подачи
Верхняя и нижняя подачи Основные понятия о государстве
Основные понятия о государстве Мой компьютер
Мой компьютер Роль системы менеджмента качества в стратегическом планировании на предприятии
Роль системы менеджмента качества в стратегическом планировании на предприятии Религиозные конфликты
Религиозные конфликты