- Изменчивость генома

Содержание
- 2. Теломераза и старение Проблемы концевой недорепликации ДНК Эффект Хейфлика
- 3. Эффект Хейфлика Монослой – нормальные соматические клетки прикрепленные ко дну сосуда, инкубируемые в термостате Контактное ингибирование-
- 4. Эффект Хейфлика Нормальные фибробласты, взятые от человеческого эмбриона, не могут делиться сколь угодно долго. Многочисленные проверки
- 5. Эффект Хейфлика Линия делящихся соматических клеток вовсе не бессмертна; старение — это свойство самих клеток (его
- 6. Эффект Хейфлика В экспериментах клетки отделяли друг от друга с помощью трипсина, а потом инкубировали в
- 7. Эффект Хейфлика а) В первоначальных опытах исходные клетки (фибробласты) получали из человеческого эмбриона - и они
- 8. Эффект Хейфлика И при переносе клеток in vitro он продолжает счет, а не начинает его сначала.
- 9. Эффект Хейфлика б) Клетки «запоминают» количество прошедших делений. «Память» об этом сохраняется и в том случае,
- 10. Эффект Хейфлика г) Важным аргументом служит изучение клеток, вошедших в фазу III. Еще за несколько делений
- 11. Эффект Хейфлика Фаза III — старение клеток in vitro. Делящимся клеткам человека и животных соответствует некий
- 12. Теломерная теория старения Теломерная теория старения (теория маргинотомии). Гипотеза «билетиков» для объяснения функционирования молекул мРНК. Последние
- 16. Теломерная теория старения При каждом делении нормальных клеток в культуре длина теломерных последовательностей сокращается на 50-100
- 17. Теломерная теория старения Укорочение теломер для клеток in vivo. В лимфоцитах по мере увеличения возраста человека
- 18. Теломерная теория старения В культивируемые клетки был введен (методами генной инженерии) ген TERT, кодирующий каталитическую субъединицу
- 19. Теломерная теория старения Обычно рассматривают два взаимно противоположных варианта: в одном случае «негатив» состоит в активации
- 20. Теломерная теория старения б) Второй возможный вариант влияния длины теломер на состояние клетки (предполагающий инактивацию «хороших»
- 21. Теломерная теория старения Теоретические предсказания А. М. Оловникова нашли экспериментальное подтверждение. а) Предсказание о том, что
- 22. Критика теломерной теории старения а) В большинстве клеток мышей - довольно высокая активность теломеразы, отчего теломерные
- 23. Критика теломерной теории старения в) В ряде случаев (кератиноциты, клетки молочной железы) введение в клетки гена
- 24. Критика теломерной теории старения Но можно ли утверждать, что старение и смерть целостного организма определяются именно
- 25. Критика теломерной теории старения б) Во-вторых, возьмем обновляющиеся ткани - например, эпидермис. В культуре эффект Хейфлика
- 26. Критика теломерной теории старения в) О том же свидетельствуют экспериментальные данные, согласно которым даже у пожилых
- 27. Критика теломерной теории старения г) Не все ясно и с самим лимитом Хейфлика. Положение о том,
- 28. Критика теломерной теории старения Начнем с того, что в организме новорожденного ребенка - порядка 10¹² всевозможных
- 29. Критика теломерной теории старения Теперь обратимся к гемопоэтическим клеткам. Среди них содержится некое количество стволовых клеток.
- 30. Критика теломерной теории старения При гомопластическом эритропоэзе основным источником эритроцитов являются деления эритробластов. Однако для поддержания
- 31. Критика теломерной теории старения Но это же означает, что, например, за 60 лет жизни человека стволовые
- 32. Критика теломерной теории старения Это приблизительные оценки, но они намного больше лимита Хейфлика, установленного для эмбриональных
- 33. Критика теломерной теории старения Этот предел (если он, действительно, существует) для разных клеток может быть совершенно
- 34. Критика теломерной теории старения а) лимит делений in vivo для различных клеток неизвестен; б) старение in
- 35. Укорочение теломер – причина старения Укорочение теломер может происходить не только в делящихся клетках - из-за
- 36. Причины старения С возрастом происходит не только укорочение теломер, но и другие изменения: увеличение числа разрывов
- 37. Теории старения Выдвинуто 2 группы теорий старения. Старение - это результат изнашивания или повреждения каких-то структур
- 38. Теории старения В организме имеется целая совокупность различных защитных систем. На внутриклеточном уровне это: система репарации
- 39. Теории старения Тогда становится понятно, почему, несмотря на эффективную систему репарации, в структуре ДНК с возрастом
- 40. Теории старения В линии половых клеток старение отсутствует Теломерная теория его полностью воспроизводит, утверждая, что в
- 41. Теории старения Первые недели эмбрионального развития, в зародыше обособляются первичные половые клетки (гоноциты); Половую дифференциацию этих
- 42. Теории старения В 1978- 1980 гг были обнаружены достоверные биохимические различия между одноименными сперматогенными клетками, полученными
- 43. Теории старения Наиболее вероятной стадией, используемой для этого, является весьма продолжительная профаза мейоза. В это время
- 44. Теории старения Принципиально возможные варианты: G - это биологический возраст клетки, т. е. некая интегральная характеристика,
- 45. Теории старения Постулат Вейсмана отражается графиком А, а в модифицированном (и более вероятном) виде - графиком
- 46. Теории старения Вариант же Е допускает, что «омоложение» одних клеток может происходить за счет других. О
- 47. Теломераза и онкогенез Кроме старения, теломеры и теломераза связаны с другой важнейшей биологической проблемой - проблемой
- 48. Теломераза и онкогенез Получение линий опухолевых клеток Нормальные соматические клетки делятся в культуре ограниченное количество раз.
- 49. Теломераза и онкогенез В обоих случаях культивировать иммортализованную линию можно опять-таки двумя способами: in vitro, т.
- 50. Теломераза и онкогенез Иммортпализация in vitro Иммортализация in vitro бывает спонтанной и индуцированной. В случае мышиных
- 51. Теломераза и онкогенез Клоны трансформантов бывают двух видов: Одни клоны имеют ограниченный пролиферативный потенциал: совершают еще
- 52. Теломераза и онкогенез В этом процессе различают две стадии: Первая стадия - временное удлинение жизни культуры,
- 53. Теломераза и онкогенез Теломеры и теломераза в трансформированных клетках Иммортализация обусловлена индукцией теломеразы - восстановление теломер
- 54. Теломераза и онкогенез Клеточные линии: а) При вирусной трансформации клеток человека на первой стадии (до кризиса)
- 55. Теломераза и онкогенез В клетках, преодолевших кризис, наблюдается увеличение длины теломер. б) Длина теломер стабилизируется за
- 56. Теломераза и онкогенез Появляется новое направление в лекарственной терапии опухолей. Следует использовать не тотальные нгибиторы синтеза
- 57. Теломераза и онкогенез г) Однако не всегда в иммортализованных клетках обнаруживается теломеразная активность. Примерно в 25
- 58. Теломераза и онкогенез д) В то же время иммортализацию ни в коем случае нельзя сводить только
- 59. Теломераза и онкогенез Удлинение теломер - лишь одно из ключевых событий иммортализации, необходимое: без него иммортализация
- 60. Теломераза и онкогенез Первичные опухоли Были протестированы на теломеразную активность несколько тысяч образцов опухолей человека. В
- 61. Метилирование ДНК Метилирование цитозина в ДНК эукариот В ядрах (и митохондриях) эукариот существуют ферменты ДНК-метилазы. Они
- 62. Метилирование ДНК Долгое время у эукариот была известна лишь одна ДНК-метилаза; она метилирует в ДНК остатки
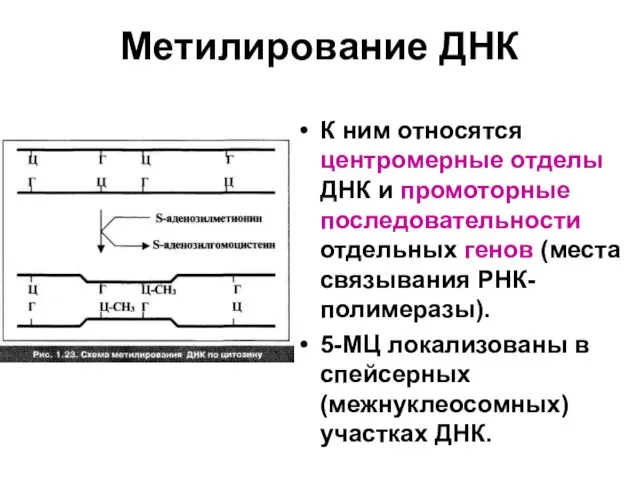
- 63. Метилирование ДНК К ним относятся центромерные отделы ДНК и промоторные последовательности отдельных генов (места связывания РНК-полимеразы).
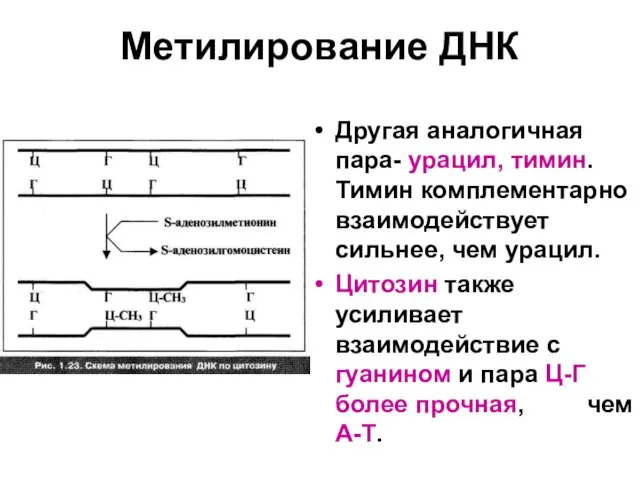
- 64. Метилирование ДНК Другая аналогичная пара- урацил, тимин. Тимин комплементарно взаимодействует сильнее, чем урацил. Цитозин также усиливает
- 65. Метилирование ДНК
- 67. Метилирование ДНК Активность ДНК-метилазы и содержание 5-МЦ в ДНК зависят от ряда обстоятельств: а) В культуре
- 68. Метилирование ДНК в) Третья параллель касается опухолевой трансформации клеток. Эта трансформация сопровождается не только появлением теломеразной
- 69. Метилирование ДНК Метилирование ДНК вовлечено в одно из начальных событий трансформации, которые обеспечивают преодоление старения и
- 70. Функции метилирования ДНК а) Участие в регуляции активности генов. Имеется положительная корреляция между функциональной активностью клеток
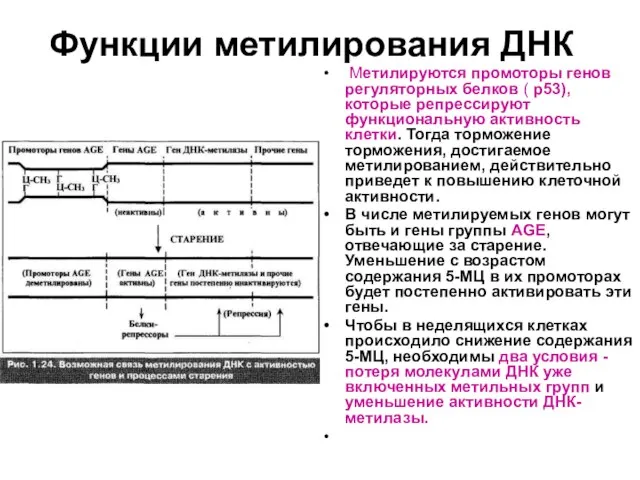
- 71. Функции метилирования ДНК Метилируются промоторы генов регуляторных белков ( р53), которые репрессируют функциональную активность клетки. Тогда
- 72. Функции метилирования ДНК Исчезновение 5-МЦ осуществляется тремя способами: истинное деметилирование, т. е. обратное превращение 5-МЦ в
- 73. Функции метилирования ДНК Диметилирование приводит к активации генов супрессорных белков, то можно предположить, что среди объектов
- 74. Функции метилирования ДНК Образуется порочный круг: недометилированность ДНК снижает активность ДНК-метилазы. Падение содержания 5-МЦ ( и
- 75. Функции метилирования ДНК В митотических клетках на баланс метильных групп влияет еще одно обстоятельство - образование
- 76. Функции метилирования ДНК При трансформации клеток ген ДНК-метилазы тем или иным способом необратимо высвобождается из-под супрессорного
- 77. Функции метилирования ДНК б) Особенно богаты 5-метилцитозином центромерные отделы ДНК. Данные отделы реплицируются не в S-фазе,
- 78. Система рестрикции и модификации у бактерий Функциональная роль метилирования ДНК у бактерий была выяснена еще в
- 79. Система рестрикции и модификации у бактерий Принцип функционирования системы Ключевая особенность бактериальных ДНК-метилаз и эндонуклеаз (рестриктаз)
- 80. Система рестрикции и модификации у бактерий При этом метилаза ДНК, узнав «свой» сайт, метилирует в нем
- 81. Система рестрикции и модификации у бактерий Проникшая в клетку чужеродная (вирусная) ДНК не защищена в этих
- 82. Действие ДНК-метилаз и рестриктаз Сразу после репликации дочерние молекулы ДНК содержат метильные группы лишь в «старых»
- 83. Система рестрикции и модификации у бактерий У бактерий метилирование собственной ДНК происходит вскоре после ее репликации
- 84. Система рестрикции и модификации у бактерий Системы типа II . сайты являются палиндромами, т. е. читаются
- 85. Система рестрикции и модификации у бактерий Именно рестриктазы типа II стали незаменимым инструментом в генно-инженерных работах.
- 86. Система рестрикции и модификации у бактерий Как же при наличии у бактерий такой защитной системы бактериофаги
- 87. Метилирование ДНК, связанное с репарацией ошибок репликации Имеется еще один вид метилирования ДНК, связанный сразу и
- 88. Метилирование ДНК, связанное с репарацией ошибок репликации Система должна распознать - какая из цепей является родительской
- 89. Метилирование ДНК, связанное с репарацией ошибок репликации После того как будет пройдено место ошибки, ферментный комплекс
- 90. Метилирование ДНК, связанное с репарацией ошибок репликации Завершает репарацию ДНК-лигаза образующая межнуклеотидную связь в месте разрыва.
- 91. Метилирование ДНК, связанное с репарацией ошибок репликации Но после того, как репликация всей ДНК и ее
- 92. Репарация повреждений ДНК В клетках происходит репарация разнообразных повреждений ДНК, постоянно появляющихся под действием всевозможных факторов.
- 93. Репарация повреждений ДНК Повреждения оснований а) Гидролитическое выщепление оснований: происходит спонтанно, а также под действием вышеперечисленных
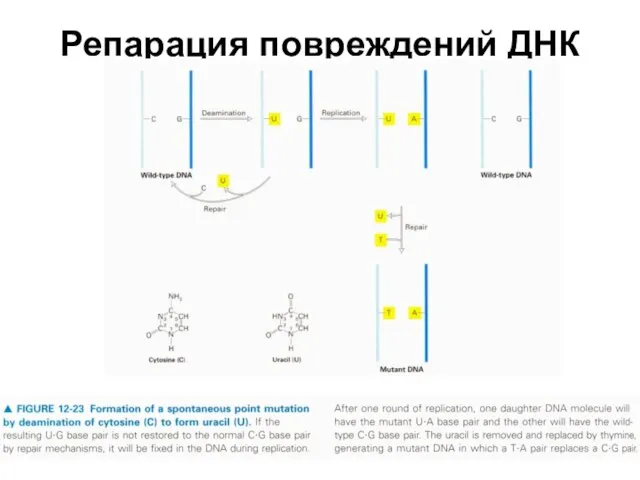
- 94. Репарация повреждений ДНК б) Гидролитическое дезаминирование оснований. В данном случае теряется не целое основание, а только
- 95. Репарация повреждений ДНК в) Образование димеров тимина. Инициируется ультрафиолетовым облучением и происходит там, где два тимидиловых
- 96. Репарация повреждений ДНК Повреждения цепей ДНК а) Одноцепочечные разрывы: между соседними нуклеотидами цепи ДНК разрывается фосфодиэфирная
- 97. Репарация повреждений ДНК б) Поперечные сшивки. Это ковалентные связи двух видов: ДНК-ДНК, т. е. между основаниями
- 98. Репарация повреждений ДНК
- 100. Примеры репарации ДНК Общий принцип репарации ДНК основан на том, что вероятность одновременного повреждения в одном
- 101. Вырезание (эксцизия) из поврежденной цепи фрагмента Фермент (эксцинуклеаза), находит место повреждения и разрывает по обе стороны
- 102. 4. Затем специальная экзонуклеаза поочередно отщепляет до 100 нуклеотидов. 5. Ресинтез фрагмента осуществляется ДНК-полимеразой Р. 6.
- 103. Примеры репарации ДНК Иногда встречаются генетические дефекты данной репарационной системы. Одна из таких наследственных болезней -
- 104. Преждевременное старение Другой вариант - синдром преждевременного старения; Это подверждает предположение о том, что и нормальное
- 105. Примеры репарации ДНК Удаление остатков урацила и репарация участков, лишенных основания 1. Остатки урацила появляются в
- 106. Примеры репарации ДНК 3. Репарационная эндонуклеаза I расщепляет остов по месту отсутствия основания. По некоторым сведениям,
- 107. Примеры репарации ДНК Однако тот же механизм не может исправлять сходное повреждение - дезаминирование 5-метилцитозина с
- 109. Скачать презентацию

Слайд 3 Эффект Хейфлика
Монослой – нормальные соматические клетки прикрепленные ко дну сосуда, инкубируемые
Эффект Хейфлика
Монослой – нормальные соматические клетки прикрепленные ко дну сосуда, инкубируемые

Слайд 4Эффект Хейфлика
Нормальные фибробласты, взятые от человеческого эмбриона, не могут делиться сколь угодно
Эффект Хейфлика
Нормальные фибробласты, взятые от человеческого эмбриона, не могут делиться сколь угодно

Слайд 5Эффект Хейфлика
Линия делящихся соматических клеток вовсе не бессмертна; старение — это свойство
Эффект Хейфлика
Линия делящихся соматических клеток вовсе не бессмертна; старение — это свойство

Слайд 6Эффект Хейфлика
В экспериментах клетки отделяли друг от друга с помощью трипсина, а
Эффект Хейфлика
В экспериментах клетки отделяли друг от друга с помощью трипсина, а

Слайд 7Эффект Хейфлика
а) В первоначальных опытах исходные клетки (фибробласты) получали из человеческого эмбриона
Эффект Хейфлика
а) В первоначальных опытах исходные клетки (фибробласты) получали из человеческого эмбриона

Слайд 8Эффект Хейфлика
И при переносе клеток in vitro он продолжает счет, а не
Эффект Хейфлика
И при переносе клеток in vitro он продолжает счет, а не

Слайд 9Эффект Хейфлика
б) Клетки «запоминают» количество прошедших делений. «Память» об этом сохраняется и
Эффект Хейфлика
б) Клетки «запоминают» количество прошедших делений. «Память» об этом сохраняется и

Слайд 10Эффект Хейфлика
г) Важным аргументом служит изучение клеток, вошедших в фазу III.
Еще за
Эффект Хейфлика
г) Важным аргументом служит изучение клеток, вошедших в фазу III.
Еще за

Слайд 11Эффект Хейфлика
Фаза III — старение клеток in vitro.
Делящимся клеткам человека и животных
Эффект Хейфлика
Фаза III — старение клеток in vitro.
Делящимся клеткам человека и животных

Слайд 12Теломерная теория старения
Теломерная теория старения (теория маргинотомии).
Гипотеза «билетиков» для объяснения функционирования
Теломерная теория старения
Теломерная теория старения (теория маргинотомии).
Гипотеза «билетиков» для объяснения функционирования


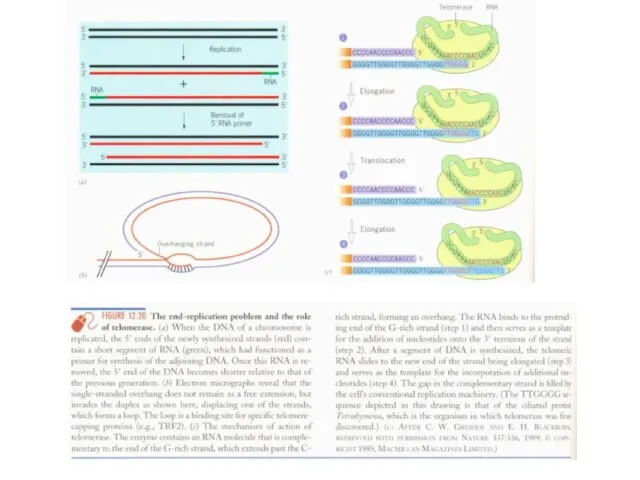
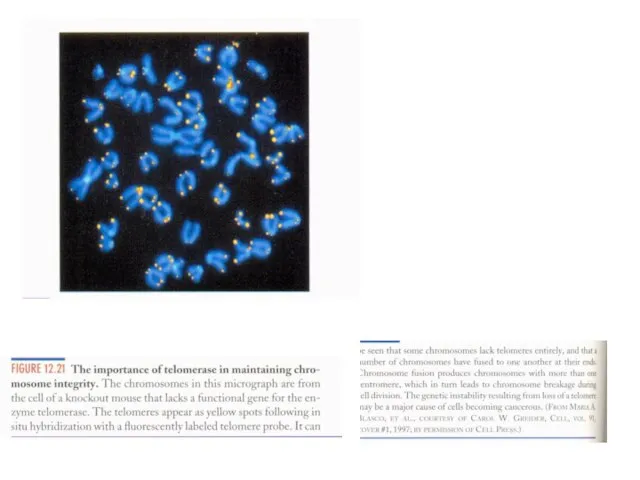
Слайд 16Теломерная теория старения
При каждом делении нормальных клеток в культуре длина теломерных последовательностей
Теломерная теория старения
При каждом делении нормальных клеток в культуре длина теломерных последовательностей

Слайд 17Теломерная теория старения
Укорочение теломер для клеток in vivo.
В лимфоцитах по мере увеличения
Теломерная теория старения
Укорочение теломер для клеток in vivo.
В лимфоцитах по мере увеличения

Слайд 18Теломерная теория старения
В культивируемые клетки был введен (методами генной инженерии) ген TERT,
Теломерная теория старения
В культивируемые клетки был введен (методами генной инженерии) ген TERT,

Слайд 19Теломерная теория старения
Обычно рассматривают два взаимно противоположных варианта:
в одном случае «негатив» состоит
Теломерная теория старения
Обычно рассматривают два взаимно противоположных варианта:
в одном случае «негатив» состоит

Слайд 20Теломерная теория старения
б) Второй возможный вариант влияния длины теломер на состояние клетки
Теломерная теория старения
б) Второй возможный вариант влияния длины теломер на состояние клетки

Слайд 21Теломерная теория старения
Теоретические предсказания А. М. Оловникова нашли экспериментальное подтверждение.
а) Предсказание о том,
Теломерная теория старения
Теоретические предсказания А. М. Оловникова нашли экспериментальное подтверждение.
а) Предсказание о том,

Слайд 22Критика теломерной теории старения
а) В большинстве клеток мышей - довольно высокая активность теломеразы,
Критика теломерной теории старения
а) В большинстве клеток мышей - довольно высокая активность теломеразы,

Слайд 23Критика теломерной теории старения
в) В ряде случаев (кератиноциты, клетки молочной железы) введение в
Критика теломерной теории старения
в) В ряде случаев (кератиноциты, клетки молочной железы) введение в

Слайд 24Критика теломерной теории старения
Но можно ли утверждать, что старение и смерть целостного
Критика теломерной теории старения
Но можно ли утверждать, что старение и смерть целостного

Слайд 25Критика теломерной теории старения
б) Во-вторых, возьмем обновляющиеся ткани - например, эпидермис.
В культуре эффект
Критика теломерной теории старения
б) Во-вторых, возьмем обновляющиеся ткани - например, эпидермис.
В культуре эффект

Слайд 26Критика теломерной теории старения
в) О том же свидетельствуют экспериментальные данные, согласно которым даже
Критика теломерной теории старения
в) О том же свидетельствуют экспериментальные данные, согласно которым даже

Слайд 27Критика теломерной теории старения
г) Не все ясно и с самим лимитом Хейфлика.
Положение о
Критика теломерной теории старения
г) Не все ясно и с самим лимитом Хейфлика. Положение о

Слайд 28Критика теломерной теории старения
Начнем с того, что в организме новорожденного ребенка -
Критика теломерной теории старения
Начнем с того, что в организме новорожденного ребенка -

Слайд 29Критика теломерной теории старения
Теперь обратимся к гемопоэтическим клеткам. Среди них содержится некое
Критика теломерной теории старения
Теперь обратимся к гемопоэтическим клеткам. Среди них содержится некое

Слайд 30Критика теломерной теории старения
При гомопластическом эритропоэзе основным источником эритроцитов являются деления эритробластов.
Критика теломерной теории старения
При гомопластическом эритропоэзе основным источником эритроцитов являются деления эритробластов.

Слайд 31Критика теломерной теории старения
Но это же означает, что, например, за 60 лет
Критика теломерной теории старения
Но это же означает, что, например, за 60 лет

Слайд 32Критика теломерной теории старения
Это приблизительные оценки, но они намного больше лимита Хейфлика,
Критика теломерной теории старения
Это приблизительные оценки, но они намного больше лимита Хейфлика,

Слайд 33Критика теломерной теории старения
Этот предел (если он, действительно, существует) для разных клеток
Критика теломерной теории старения
Этот предел (если он, действительно, существует) для разных клеток

Слайд 34Критика теломерной теории старения
а) лимит делений in vivo для различных клеток неизвестен;
б) старение in
Критика теломерной теории старения
а) лимит делений in vivo для различных клеток неизвестен;
б) старение in

Слайд 35Укорочение теломер – причина старения
Укорочение теломер может происходить не только в
Укорочение теломер – причина старения
Укорочение теломер может происходить не только в
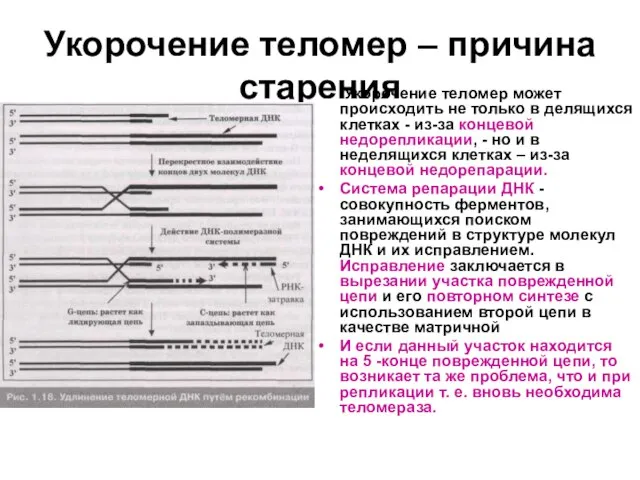
Слайд 36Причины старения
С возрастом происходит не только укорочение теломер, но и другие
Причины старения
С возрастом происходит не только укорочение теломер, но и другие

Слайд 37Теории старения
Выдвинуто 2 группы теорий старения.
Старение - это результат изнашивания
Теории старения
Выдвинуто 2 группы теорий старения.
Старение - это результат изнашивания

Слайд 38Теории старения
В организме имеется целая совокупность различных защитных систем. На внутриклеточном уровне
Теории старения
В организме имеется целая совокупность различных защитных систем. На внутриклеточном уровне

Слайд 39Теории старения
Тогда становится понятно, почему, несмотря на эффективную систему репарации, в структуре
Теории старения
Тогда становится понятно, почему, несмотря на эффективную систему репарации, в структуре

Слайд 40Теории старения
В линии половых клеток старение отсутствует
Теломерная теория его полностью воспроизводит,
Теории старения
В линии половых клеток старение отсутствует
Теломерная теория его полностью воспроизводит,

Слайд 41Теории старения
Первые недели эмбрионального развития, в зародыше обособляются первичные половые клетки (гоноциты);
Половую
Теории старения
Первые недели эмбрионального развития, в зародыше обособляются первичные половые клетки (гоноциты);
Половую

Слайд 42Теории старения
В 1978- 1980 гг были обнаружены достоверные биохимические различия между одноименными
Теории старения
В 1978- 1980 гг были обнаружены достоверные биохимические различия между одноименными

Слайд 43Теории старения
Наиболее вероятной стадией, используемой для этого, является весьма продолжительная профаза мейоза.
Теории старения
Наиболее вероятной стадией, используемой для этого, является весьма продолжительная профаза мейоза.

Слайд 44Теории старения
Принципиально возможные варианты:
G - это биологический возраст клетки, т. е.
Теории старения
Принципиально возможные варианты:
G - это биологический возраст клетки, т. е.
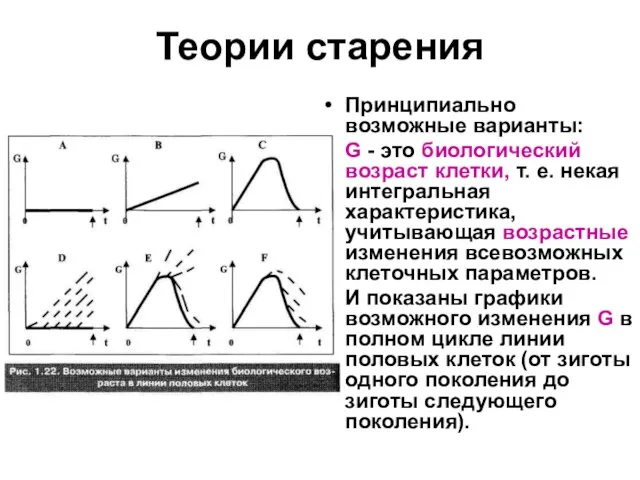
Слайд 45Теории старения
Постулат Вейсмана отражается графиком А, а в модифицированном (и более вероятном)
Теории старения
Постулат Вейсмана отражается графиком А, а в модифицированном (и более вероятном)
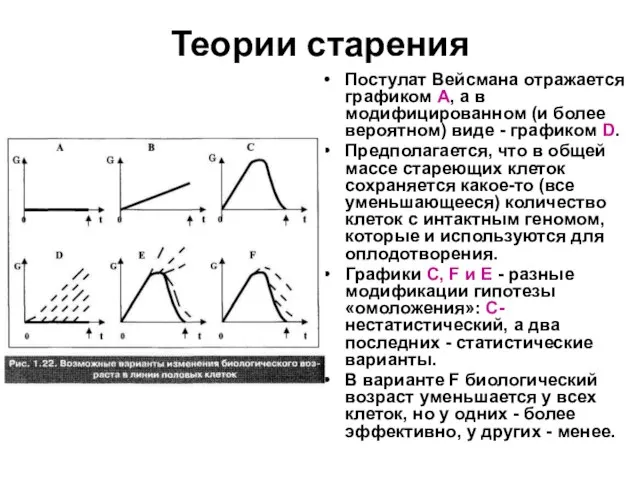
Слайд 46Теории старения
Вариант же Е допускает, что «омоложение» одних клеток может происходить за
Теории старения
Вариант же Е допускает, что «омоложение» одних клеток может происходить за
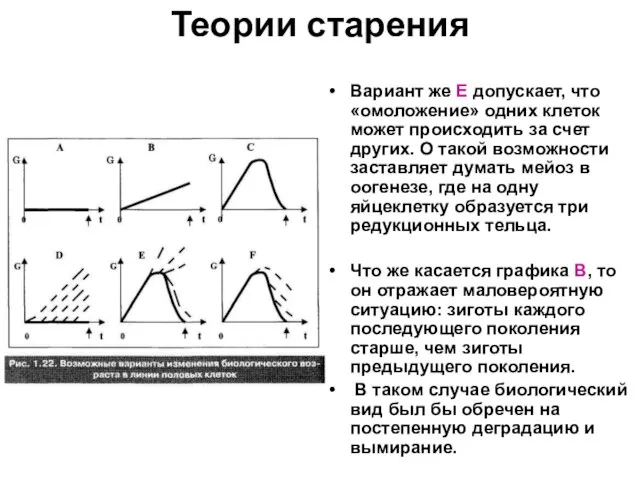
Слайд 47Теломераза и онкогенез
Кроме старения, теломеры и теломераза связаны с другой важнейшей биологической
Теломераза и онкогенез
Кроме старения, теломеры и теломераза связаны с другой важнейшей биологической

Слайд 48Теломераза и онкогенез
Получение линий опухолевых клеток
Нормальные соматические клетки делятся в культуре
Теломераза и онкогенез
Получение линий опухолевых клеток
Нормальные соматические клетки делятся в культуре

Слайд 49Теломераза и онкогенез
В обоих случаях культивировать иммортализованную линию можно опять-таки двумя способами:
in
Теломераза и онкогенез
В обоих случаях культивировать иммортализованную линию можно опять-таки двумя способами:
in

Слайд 50Теломераза и онкогенез
Иммортпализация in vitro
Иммортализация in vitro бывает спонтанной и индуцированной.
В
Теломераза и онкогенез
Иммортпализация in vitro
Иммортализация in vitro бывает спонтанной и индуцированной.
В

Слайд 51Теломераза и онкогенез
Клоны трансформантов бывают двух видов:
Одни клоны имеют ограниченный пролиферативный
Теломераза и онкогенез
Клоны трансформантов бывают двух видов:
Одни клоны имеют ограниченный пролиферативный

Слайд 52Теломераза и онкогенез
В этом процессе различают две стадии:
Первая стадия - временное
Теломераза и онкогенез
В этом процессе различают две стадии:
Первая стадия - временное

Слайд 53Теломераза и онкогенез
Теломеры и теломераза в трансформированных клетках
Иммортализация обусловлена индукцией теломеразы -
Теломераза и онкогенез
Теломеры и теломераза в трансформированных клетках
Иммортализация обусловлена индукцией теломеразы -

Слайд 54Теломераза и онкогенез
Клеточные линии:
а) При вирусной трансформации клеток человека на первой стадии
Теломераза и онкогенез
Клеточные линии:
а) При вирусной трансформации клеток человека на первой стадии

Слайд 55Теломераза и онкогенез
В клетках, преодолевших кризис, наблюдается увеличение длины теломер.
б) Длина
Теломераза и онкогенез
В клетках, преодолевших кризис, наблюдается увеличение длины теломер.
б) Длина

Слайд 56Теломераза и онкогенез
Появляется новое направление в лекарственной терапии опухолей. Следует использовать не
Теломераза и онкогенез
Появляется новое направление в лекарственной терапии опухолей. Следует использовать не

Слайд 57Теломераза и онкогенез
г) Однако не всегда в иммортализованных клетках обнаруживается теломеразная активность.
Примерно
Теломераза и онкогенез
г) Однако не всегда в иммортализованных клетках обнаруживается теломеразная активность.
Примерно

Слайд 58Теломераза и онкогенез
д) В то же время иммортализацию ни в коем случае
Теломераза и онкогенез
д) В то же время иммортализацию ни в коем случае

Слайд 59Теломераза и онкогенез
Удлинение теломер - лишь одно из ключевых событий иммортализации, необходимое:
Теломераза и онкогенез
Удлинение теломер - лишь одно из ключевых событий иммортализации, необходимое:

Слайд 60Теломераза и онкогенез
Первичные опухоли
Были протестированы на теломеразную активность несколько тысяч образцов
Теломераза и онкогенез
Первичные опухоли
Были протестированы на теломеразную активность несколько тысяч образцов

Слайд 61Метилирование ДНК
Метилирование цитозина в ДНК эукариот
В ядрах (и митохондриях) эукариот существуют
Метилирование ДНК
Метилирование цитозина в ДНК эукариот
В ядрах (и митохондриях) эукариот существуют
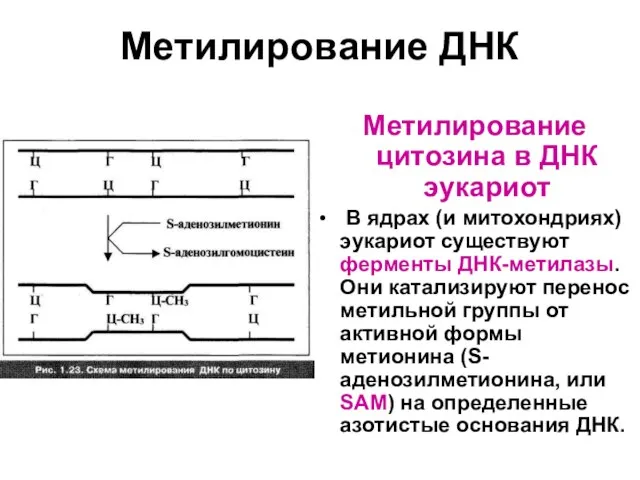
Слайд 62Метилирование ДНК
Долгое время у эукариот была известна лишь одна ДНК-метилаза; она метилирует
Метилирование ДНК
Долгое время у эукариот была известна лишь одна ДНК-метилаза; она метилирует
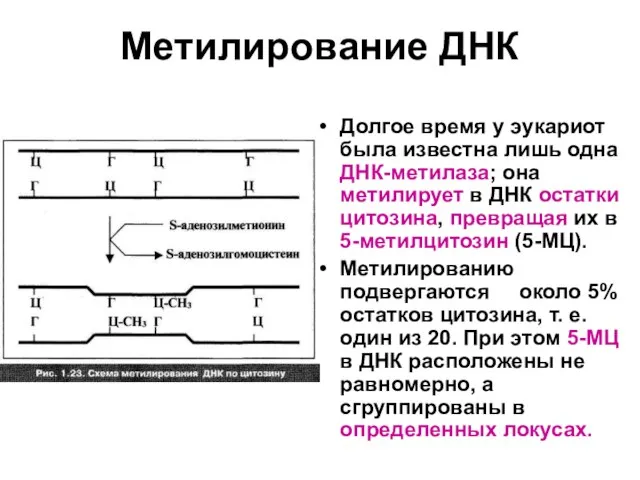
Слайд 63Метилирование ДНК
К ним относятся центромерные отделы ДНК и промоторные последовательности отдельных генов
Метилирование ДНК
К ним относятся центромерные отделы ДНК и промоторные последовательности отдельных генов
Слайд 64Метилирование ДНК
Другая аналогичная пара- урацил, тимин. Тимин комплементарно взаимодействует сильнее, чем урацил.
Цитозин
Метилирование ДНК
Другая аналогичная пара- урацил, тимин. Тимин комплементарно взаимодействует сильнее, чем урацил.
Цитозин
Слайд 65Метилирование ДНК
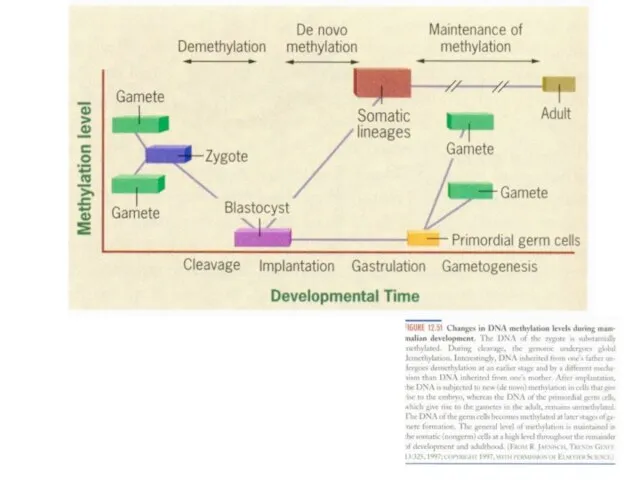
Метилирование ДНК

Слайд 67Метилирование ДНК
Активность ДНК-метилазы и содержание 5-МЦ в ДНК зависят от ряда
Метилирование ДНК
Активность ДНК-метилазы и содержание 5-МЦ в ДНК зависят от ряда

Слайд 68Метилирование ДНК
в) Третья параллель касается опухолевой трансформации клеток. Эта трансформация сопровождается не
Метилирование ДНК
в) Третья параллель касается опухолевой трансформации клеток. Эта трансформация сопровождается не

Слайд 69Метилирование ДНК
Метилирование ДНК вовлечено в одно из начальных событий трансформации, которые
Метилирование ДНК
Метилирование ДНК вовлечено в одно из начальных событий трансформации, которые

Слайд 70Функции метилирования ДНК
а) Участие в регуляции активности генов. Имеется положительная корреляция между
Функции метилирования ДНК
а) Участие в регуляции активности генов. Имеется положительная корреляция между

Слайд 71Функции метилирования ДНК
Метилируются промоторы генов регуляторных белков ( р53), которые репрессируют
Функции метилирования ДНК
Метилируются промоторы генов регуляторных белков ( р53), которые репрессируют
Слайд 72Функции метилирования ДНК
Исчезновение 5-МЦ осуществляется тремя способами:
истинное деметилирование, т. е. обратное превращение
Функции метилирования ДНК
Исчезновение 5-МЦ осуществляется тремя способами:
истинное деметилирование, т. е. обратное превращение

Слайд 73Функции метилирования ДНК
Диметилирование приводит к активации генов супрессорных белков, то можно предположить,
Функции метилирования ДНК
Диметилирование приводит к активации генов супрессорных белков, то можно предположить,
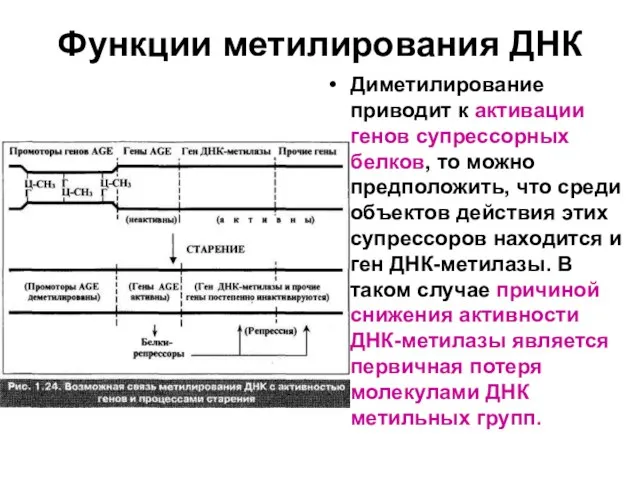
Слайд 74Функции метилирования ДНК
Образуется порочный круг: недометилированность ДНК снижает активность ДНК-метилазы.
Падение содержания
Функции метилирования ДНК
Образуется порочный круг: недометилированность ДНК снижает активность ДНК-метилазы.
Падение содержания
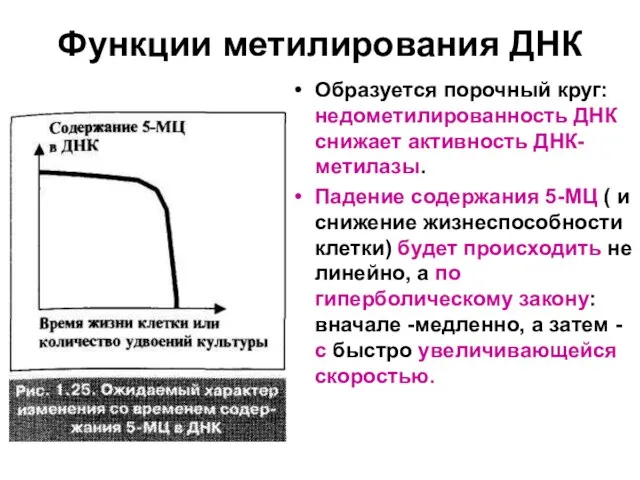
Слайд 75Функции метилирования ДНК
В митотических клетках на баланс метильных групп влияет еще одно
Функции метилирования ДНК
В митотических клетках на баланс метильных групп влияет еще одно
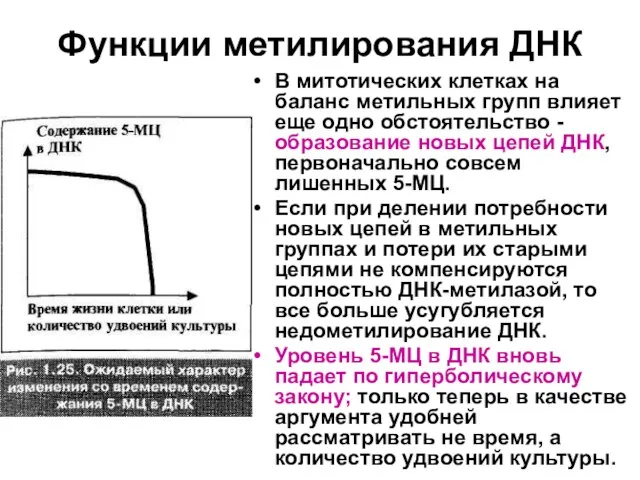
Слайд 76Функции метилирования ДНК
При трансформации клеток ген ДНК-метилазы тем или иным способом необратимо
Функции метилирования ДНК
При трансформации клеток ген ДНК-метилазы тем или иным способом необратимо

Слайд 77Функции метилирования ДНК
б) Особенно богаты 5-метилцитозином центромерные отделы ДНК. Данные отделы
Функции метилирования ДНК
б) Особенно богаты 5-метилцитозином центромерные отделы ДНК. Данные отделы

Слайд 78Система рестрикции и модификации у бактерий
Функциональная роль метилирования ДНК у бактерий
Система рестрикции и модификации у бактерий
Функциональная роль метилирования ДНК у бактерий

Слайд 79Система рестрикции и модификации у бактерий
Принцип функционирования системы
Ключевая особенность бактериальных ДНК-метилаз
Система рестрикции и модификации у бактерий
Принцип функционирования системы
Ключевая особенность бактериальных ДНК-метилаз

Слайд 80Система рестрикции и модификации у бактерий
При этом метилаза ДНК, узнав «свой» сайт,
Система рестрикции и модификации у бактерий
При этом метилаза ДНК, узнав «свой» сайт,

Слайд 81Система рестрикции и модификации у бактерий
Проникшая в клетку чужеродная (вирусная) ДНК не
Система рестрикции и модификации у бактерий
Проникшая в клетку чужеродная (вирусная) ДНК не

Слайд 82Действие ДНК-метилаз и рестриктаз
Сразу после репликации дочерние молекулы ДНК содержат метильные группы
Действие ДНК-метилаз и рестриктаз
Сразу после репликации дочерние молекулы ДНК содержат метильные группы

Слайд 83Система рестрикции и модификации у бактерий
У бактерий метилирование собственной ДНК происходит вскоре
Система рестрикции и модификации у бактерий
У бактерий метилирование собственной ДНК происходит вскоре

Слайд 84Система рестрикции и модификации у бактерий
Системы типа II .
сайты являются палиндромами,
Система рестрикции и модификации у бактерий
Системы типа II .
сайты являются палиндромами,

Слайд 85Система рестрикции и модификации у бактерий
Именно рестриктазы типа II стали незаменимым
Система рестрикции и модификации у бактерий
Именно рестриктазы типа II стали незаменимым

Слайд 86Система рестрикции и модификации у бактерий
Как же при наличии у бактерий
Система рестрикции и модификации у бактерий
Как же при наличии у бактерий

Слайд 87Метилирование ДНК, связанное с репарацией ошибок репликации
Имеется еще один вид метилирования ДНК,
Метилирование ДНК, связанное с репарацией ошибок репликации
Имеется еще один вид метилирования ДНК,

Слайд 88 Метилирование ДНК, связанное с репарацией ошибок репликации
Система должна распознать - какая
Метилирование ДНК, связанное с репарацией ошибок репликации
Система должна распознать - какая

Слайд 89 Метилирование ДНК, связанное с репарацией ошибок репликации
После того как будет пройдено
Метилирование ДНК, связанное с репарацией ошибок репликации
После того как будет пройдено

Слайд 90Метилирование ДНК, связанное с репарацией ошибок репликации
Завершает репарацию ДНК-лигаза образующая межнуклеотидную связь
Метилирование ДНК, связанное с репарацией ошибок репликации
Завершает репарацию ДНК-лигаза образующая межнуклеотидную связь

Слайд 91Метилирование ДНК, связанное с репарацией ошибок репликации
Но после того, как репликация всей
Метилирование ДНК, связанное с репарацией ошибок репликации
Но после того, как репликация всей

Слайд 92Репарация повреждений ДНК
В клетках происходит репарация разнообразных повреждений ДНК, постоянно появляющихся
Репарация повреждений ДНК
В клетках происходит репарация разнообразных повреждений ДНК, постоянно появляющихся

Слайд 93Репарация повреждений ДНК
Повреждения оснований
а) Гидролитическое выщепление оснований: происходит
спонтанно, а также под действием
Репарация повреждений ДНК
Повреждения оснований
а) Гидролитическое выщепление оснований: происходит
спонтанно, а также под действием

Слайд 94Репарация повреждений ДНК
б) Гидролитическое дезаминирование оснований. В данном случае теряется не целое основание,
Репарация повреждений ДНК
б) Гидролитическое дезаминирование оснований. В данном случае теряется не целое основание,

Слайд 95Репарация повреждений ДНК
в) Образование димеров тимина. Инициируется ультрафиолетовым облучением и происходит там, где
Репарация повреждений ДНК
в) Образование димеров тимина. Инициируется ультрафиолетовым облучением и происходит там, где

Слайд 96Репарация повреждений ДНК
Повреждения цепей ДНК
а) Одноцепочечные разрывы: между соседними нуклеотидами цепи ДНК
Репарация повреждений ДНК
Повреждения цепей ДНК
а) Одноцепочечные разрывы: между соседними нуклеотидами цепи ДНК

Слайд 97Репарация повреждений ДНК
б) Поперечные сшивки. Это ковалентные связи двух видов:
ДНК-ДНК, т. е.
Репарация повреждений ДНК
б) Поперечные сшивки. Это ковалентные связи двух видов:
ДНК-ДНК, т. е.

Слайд 98Репарация повреждений ДНК
Репарация повреждений ДНК
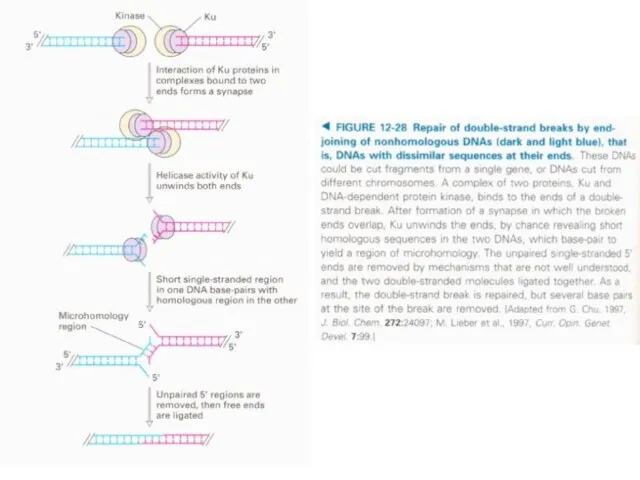
Слайд 100Примеры репарации ДНК
Общий принцип репарации ДНК основан на том, что вероятность одновременного
Примеры репарации ДНК
Общий принцип репарации ДНК основан на том, что вероятность одновременного

Слайд 101Вырезание (эксцизия) из поврежденной цепи фрагмента
Фермент (эксцинуклеаза), находит место повреждения и
разрывает
Вырезание (эксцизия) из поврежденной цепи фрагмента
Фермент (эксцинуклеаза), находит место повреждения и
разрывает
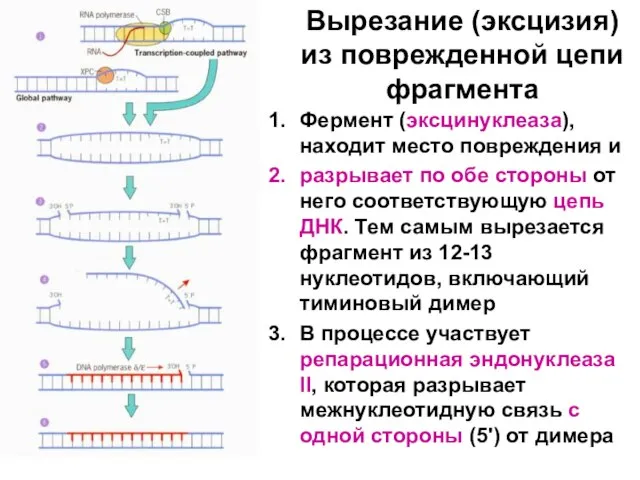
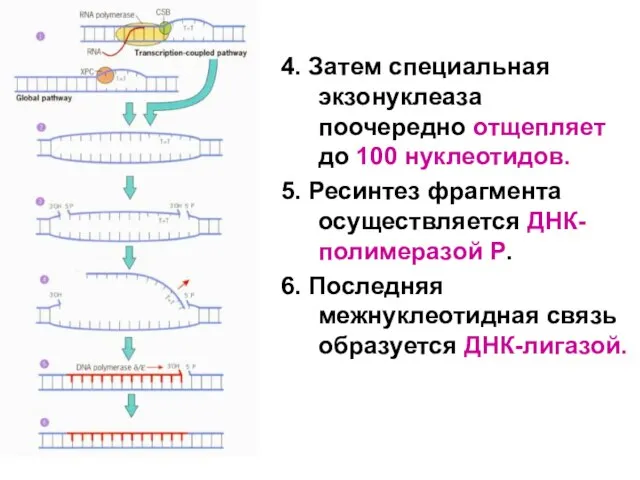
Слайд 1024. Затем специальная экзонуклеаза поочередно отщепляет до 100 нуклеотидов.
5. Ресинтез фрагмента осуществляется
4. Затем специальная экзонуклеаза поочередно отщепляет до 100 нуклеотидов.
5. Ресинтез фрагмента осуществляется
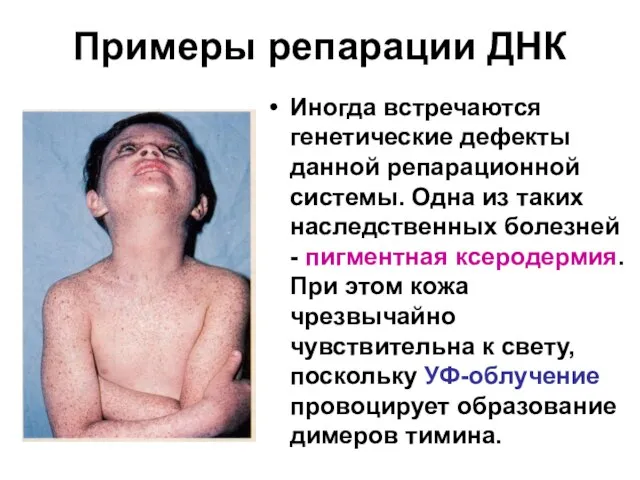
Слайд 103Примеры репарации ДНК
Иногда встречаются генетические дефекты данной репарационной системы. Одна из таких
Примеры репарации ДНК
Иногда встречаются генетические дефекты данной репарационной системы. Одна из таких
Слайд 104Преждевременное старение
Другой вариант - синдром преждевременного старения;
Это подверждает предположение о том,
Преждевременное старение
Другой вариант - синдром преждевременного старения;
Это подверждает предположение о том,

Слайд 105Примеры репарации ДНК
Удаление остатков урацила и репарация участков, лишенных основания
1. Остатки
Примеры репарации ДНК
Удаление остатков урацила и репарация участков, лишенных основания
1. Остатки

Слайд 106Примеры репарации ДНК
3. Репарационная эндонуклеаза I расщепляет остов по месту отсутствия основания.
Примеры репарации ДНК
3. Репарационная эндонуклеаза I расщепляет остов по месту отсутствия основания.

Слайд 107Примеры репарации ДНК
Однако тот же механизм не может исправлять сходное повреждение -
Примеры репарации ДНК
Однако тот же механизм не может исправлять сходное повреждение -

 Тема 1. Войны и военные конфликты Занятие 1. Понятие, виды войн и военных конфликтов. Классификация войн. Специфика и характер совре
Тема 1. Войны и военные конфликты Занятие 1. Понятие, виды войн и военных конфликтов. Классификация войн. Специфика и характер совре Презентация на тему Задача таможенного менеджмента как теории
Презентация на тему Задача таможенного менеджмента как теории Потолочная плитка
Потолочная плитка Комитетпо event-менеджментупри АКМР
Комитетпо event-менеджментупри АКМР Приготовление блюда из варёных и припущенных овощей
Приготовление блюда из варёных и припущенных овощей День народного единства
День народного единства Курсовая работа
Курсовая работа Программно-аппаратный комплекс для построения среды электронного документооборота
Программно-аппаратный комплекс для построения среды электронного документооборота Политическая система Италии
Политическая система Италии Великие озера Северной Америки
Великие озера Северной Америки The Gerund Герундий
The Gerund Герундий Введение в организацию белковой структуры и молекулярное моделирование
Введение в организацию белковой структуры и молекулярное моделирование Плакаты в годы войны
Плакаты в годы войны 10190_7807306_13
10190_7807306_13 Части речи Урок-презентация по русскому языку. Выполнила учитель начальных классов Липушкина Елена Васильевна Липушкина Елена Ва
Части речи Урок-презентация по русскому языку. Выполнила учитель начальных классов Липушкина Елена Васильевна Липушкина Елена Ва Ученическое самоуправление МОУ СОШ №3 с.п. Аргудан
Ученическое самоуправление МОУ СОШ №3 с.п. Аргудан Социальный проект «Green Town» «Зеленый город» Авторы проекта: учащиеся МОУ СОШ №15 Руководитель: Шагалова Елена Михайловна
Социальный проект «Green Town» «Зеленый город» Авторы проекта: учащиеся МОУ СОШ №15 Руководитель: Шагалова Елена Михайловна Московский городской психолого-педагогический университет
Московский городской психолого-педагогический университет ДЕЯТЕЛЬНОСТНЫЙ ПОДХОД НА УРОКАХ РУССКОГО ЯЗЫКА В 1 КЛАССЕ
ДЕЯТЕЛЬНОСТНЫЙ ПОДХОД НА УРОКАХ РУССКОГО ЯЗЫКА В 1 КЛАССЕ Исследовательская группа №6
Исследовательская группа №6 Бесконечно убывающая геометрическая прогрессия_. (10 класс)
Бесконечно убывающая геометрическая прогрессия_. (10 класс) Презентация к диплому
Презентация к диплому СПРЕЙ КОМПЛЕКСНОГО ДЕЙСТВИЯ ДЛЯ НОСА
СПРЕЙ КОМПЛЕКСНОГО ДЕЙСТВИЯ ДЛЯ НОСА Проектная деятельность учащихся
Проектная деятельность учащихся СПИД - наиболее опасное инфекционное заболевание.
СПИД - наиболее опасное инфекционное заболевание. dc687bf20b1e430dbaadc8c0ccada226
dc687bf20b1e430dbaadc8c0ccada226 Отель Sunset-hill г.Адлер
Отель Sunset-hill г.Адлер Сравнение умственной работоспособности обучающихся младшей и старшей школы в стрессовой ситуации
Сравнение умственной работоспособности обучающихся младшей и старшей школы в стрессовой ситуации