- Первичные иммунодефициты у взрослых: проблемы диагностики и лечения

Содержание
- 2. Иммунитет есть совокупность биологических явлений и процессов, направленных на сохранение постоянства внутренней среды организма от генетически
- 3. Что же такое иммунодефицит? Состояние, при котором иммунная система неспособна выполнять свои нормальные функции, а именно
- 4. Клинические ключи к диагностике иммунодефицита 2. Основные клинические признаки 2.1. Частые простудные заболевания (более 4 раз
- 5. Заболевания, при которых повышен риск инфекции
- 6. Диагноз иммунодефицита подразумевает исключение иных причин, способствующих развитию инфекционного процесса Например: Рецидив инфекционного процесса в одном
- 7. Клинический алгоритм диагностики и лечения иммунодефицитов I. НАЛИЧИЕ ИММУНОДЕФИЦИТА 1. Имеется 2. Отсутствует II. УРОВЕНЬ ЛОКАЛИЗАЦИИ
- 8. Классификация иммунодефицитов
- 9. Что лежит в основе иммунодефицита
- 10. Что страдает при иммунодефицитах
- 11. Классификация первичных иммунодефицитов В соответствие с классификацией ВОЗ (IUIS, International Union of Immunological Societies, affiliated to
- 12. Нарушение звеньев иммунитета при первичных иммунодефицитах Нарушения клеточного иммунитета:(иммунодефициты, связанные большими наследственными дефектами) синдром Ди Джорджи
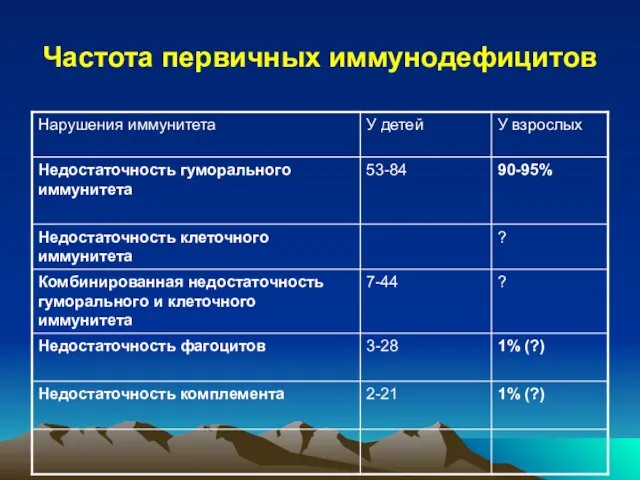
- 13. Частота первичных иммунодефицитов
- 14. Механизмы компенсации Многоуровневый характер иммунной системы Многократное дублирование отдельных иммунных реакций Успешное лечение первичных иммунодефицитов в
- 15. Причины вторичных иммунодефицитов Проявление первичных иммунодефицитов Диагностика возможна в более позднем возрасте
- 16. Иммунодефициты с преимущественным поражением гуморального звена
- 17. Общий вариабельный иммунодефицит Причины: распространенность у детей – 1:25000-66000, у взрослых – неизвестна; у большинства диагностируется
- 18. Клинические особенности первичные иммунодефицитов с преимущественным поражением гуморального звена Хронические воспалительные заболевания верхних и нижних дыхательных
- 19. Диагноз, лечение Подозрение на общую вариабельную иммунную недостаточность (ОВИН) возникает при обследовании детей или взрослых с
- 20. Селективный дефицит подклассов IgG IgG 1 (70%) - основной реактант при формировании антител против белковых антигенов
- 21. Селективный дефицит подклассов иммуноглобулинов Причины: Впервые описан в 1970г.; точная частота иммунодефицита неизвестна, но может достигать
- 22. Дефицит специфического иммунного ответа при нормальном уровне иммуноглобулинов Причины: вероятно, что он встречается гораздо чаще, чем
- 23. Нарушение иммунного ответа на полисахаридные антигены Причины: Характеризуется рецидивирующими бактериальными инфекциями при нормальном ответе на белковые
- 24. Селективный дефицит IgA Причины: самый частый иммунодефицит (1:500), но самый незаметный; причины неизвестны; может быть ассоциирован
- 25. Гипер-IgM Причины: Возникает в результате Т-клеточного дефекта, но клинические проявления как при гуморальном иммунодефиците (характерно для
- 26. Гипер-IgE-синдром (синдром Джоба) Причины: частота неизвестна, механизмы вероятно связаны с нарушением функции Т- и В-лимфоцитов; в
- 27. Комбинированные иммунодефициты
- 28. Особенности клинических проявлений вторичных иммунодефицитов при недостаточности клеточного иммунитета Клинические проявления с первых недель/месяцев жизни Возбудителями
- 29. У взрослых Первичные комбинированные иммунодефициты и иммунодефициты по клеточному звену практически не встречаются
- 30. Синдром Гуда (тимома с наличием иммунодефицита) Причины: редкий комбинированный В- и Т-клеточный первичный иммунодефицит у взрослых,
- 31. Синдром Гуда (тимома с наличием иммунодефицита) Клиника: часто бессимптомно, тимома диагностируется случайно (при рентгенографии грудной клетки
- 32. Синдром Гуда (тимома с наличием иммунодефицита) Лечение: оперативное лечение, постоянная заместительная терапия внутривенным введением иммуноглобулинов для
- 33. Недостаточность фагоцитарного звена
- 34. Особенности клинических проявлений при нарушении неспецифического иммунитета Длительно текущие и рецидивирующие инфекции Скудность клинических проявлений, несмотря
- 35. Хроническая гранулематозная болезнь (ХГБ) Причины: имеется дефект внутриклеточного бактериального киллинга (нарушено образование супероксида, кислородных радикалов и
- 36. Циклическая нейтропения Причина: Циклическое изменение количества нейтрофилов; цикл обычно 21 день Клиника: язвы околоротовой полости (на
- 37. Патология системы комплемента
- 38. Особенности клинических при нарушении в системе комплимента C1, C2, C4 D, B, P C3 C5, C6,
- 39. Наследственный ангионевротический отек Причины: аутосомно-доминантное тип, характеризующийся снижением активности ингибитора С1-эстеразу, что ведет к быстрой неконтролируемой
- 40. Иммуностимуляторы, применяемые для лечения первичных иимунодефицитов Иммуноглобулины: иммуноглобулин человека нормальный для в/в введения, хумоглобин, интраглобин, пентаглобин,
- 41. СПАСИБО
- 42. Клинико-анамнестические признаки иммунодефицита Частые простудные заболевания (более 4 раз в год) Рецидивирующий герпес (4-6 раз в
- 43. Ключи к диагностике ПЕРВИЧНОГО иммунодефицита 1. Возраст до 5 лет 2. Главные* 2.1.Частые заболевания отитом (не
- 44. Ключи к диагностике ВТОРИЧНОГО иммунодефицита 1. В анамнезе 1.1.Вирусные инфекции (HIV, EBV, CMV, вирусы краснухи, энтеровирусы
- 45. Ключи к диагностике ВТОРИЧНОГО иммунодефицита 2. Главные клинические признаки 2.1. Частые простудные заболевания (более 4 раз
- 47. Скачать презентацию
Слайд 2Иммунитет есть совокупность биологических явлений и процессов, направленных на сохранение постоянства внутренней
Иммунитет есть совокупность биологических явлений и процессов, направленных на сохранение постоянства внутренней

Слайд 3Что же такое иммунодефицит?
Состояние, при котором иммунная система неспособна выполнять свои нормальные
Что же такое иммунодефицит?
Состояние, при котором иммунная система неспособна выполнять свои нормальные

Слайд 4Клинические ключи к диагностике иммунодефицита
2. Основные клинические признаки
2.1. Частые простудные заболевания (более
Клинические ключи к диагностике иммунодефицита
2. Основные клинические признаки
2.1. Частые простудные заболевания (более

Слайд 5Заболевания, при которых повышен риск инфекции
Заболевания, при которых повышен риск инфекции

Слайд 6Диагноз иммунодефицита подразумевает исключение иных причин, способствующих развитию инфекционного процесса
Например:
Рецидив инфекционного процесса
Диагноз иммунодефицита подразумевает исключение иных причин, способствующих развитию инфекционного процесса
Например:
Рецидив инфекционного процесса

Слайд 7Клинический алгоритм диагностики и лечения иммунодефицитов
I. НАЛИЧИЕ ИММУНОДЕФИЦИТА
1. Имеется
2. Отсутствует
II. УРОВЕНЬ ЛОКАЛИЗАЦИИ
Клинический алгоритм диагностики и лечения иммунодефицитов
I. НАЛИЧИЕ ИММУНОДЕФИЦИТА
1. Имеется
2. Отсутствует
II. УРОВЕНЬ ЛОКАЛИЗАЦИИ

Слайд 8Классификация иммунодефицитов
Классификация иммунодефицитов

Слайд 9Что лежит в основе иммунодефицита
Что лежит в основе иммунодефицита

Слайд 10Что страдает при иммунодефицитах
Что страдает при иммунодефицитах

Слайд 11Классификация первичных иммунодефицитов
В соответствие с классификацией ВОЗ (IUIS, International Union of Immunological
Классификация первичных иммунодефицитов
В соответствие с классификацией ВОЗ (IUIS, International Union of Immunological

Слайд 12Нарушение звеньев иммунитета при первичных иммунодефицитах
Нарушения клеточного иммунитета:(иммунодефициты, связанные большими наследственными дефектами)
Нарушение звеньев иммунитета при первичных иммунодефицитах
Нарушения клеточного иммунитета:(иммунодефициты, связанные большими наследственными дефектами)

Слайд 13Частота первичных иммунодефицитов
Частота первичных иммунодефицитов
Слайд 14Механизмы компенсации
Многоуровневый характер иммунной системы
Многократное дублирование отдельных иммунных реакций
Успешное лечение первичных иммунодефицитов
Механизмы компенсации
Многоуровневый характер иммунной системы
Многократное дублирование отдельных иммунных реакций
Успешное лечение первичных иммунодефицитов

Слайд 15Причины вторичных иммунодефицитов
Проявление первичных иммунодефицитов
Диагностика возможна в более позднем возрасте
Причины вторичных иммунодефицитов
Проявление первичных иммунодефицитов
Диагностика возможна в более позднем возрасте

Слайд 16Иммунодефициты с преимущественным поражением гуморального звена
Иммунодефициты с преимущественным поражением гуморального звена

Слайд 17Общий вариабельный иммунодефицит
Причины: распространенность у детей – 1:25000-66000, у взрослых – неизвестна;
Общий вариабельный иммунодефицит
Причины: распространенность у детей – 1:25000-66000, у взрослых – неизвестна;

Слайд 18Клинические особенности первичные иммунодефицитов с преимущественным поражением гуморального звена
Хронические воспалительные заболевания верхних
Клинические особенности первичные иммунодефицитов с преимущественным поражением гуморального звена
Хронические воспалительные заболевания верхних

Слайд 19Диагноз, лечение
Подозрение на общую вариабельную иммунную недостаточность (ОВИН) возникает при обследовании детей
Диагноз, лечение
Подозрение на общую вариабельную иммунную недостаточность (ОВИН) возникает при обследовании детей

Слайд 20Селективный дефицит подклассов IgG
IgG 1 (70%) - основной реактант при формировании антител
Селективный дефицит подклассов IgG
IgG 1 (70%) - основной реактант при формировании антител

Слайд 21Селективный дефицит подклассов иммуноглобулинов
Причины:
Впервые описан в 1970г.; точная частота иммунодефицита неизвестна, но
Селективный дефицит подклассов иммуноглобулинов
Причины:
Впервые описан в 1970г.; точная частота иммунодефицита неизвестна, но

Слайд 22Дефицит специфического иммунного ответа при нормальном уровне иммуноглобулинов
Причины: вероятно, что он встречается
Дефицит специфического иммунного ответа при нормальном уровне иммуноглобулинов
Причины: вероятно, что он встречается

Слайд 23Нарушение иммунного ответа на полисахаридные антигены
Причины: Характеризуется рецидивирующими бактериальными инфекциями при нормальном
Нарушение иммунного ответа на полисахаридные антигены
Причины: Характеризуется рецидивирующими бактериальными инфекциями при нормальном

Слайд 24Селективный дефицит IgA
Причины:
самый частый иммунодефицит (1:500), но самый незаметный; причины неизвестны; может
Селективный дефицит IgA
Причины:
самый частый иммунодефицит (1:500), но самый незаметный; причины неизвестны; может

Слайд 25Гипер-IgM
Причины: Возникает в результате Т-клеточного дефекта, но клинические проявления как при гуморальном
Гипер-IgM
Причины: Возникает в результате Т-клеточного дефекта, но клинические проявления как при гуморальном

Слайд 26Гипер-IgE-синдром (синдром Джоба)
Причины: частота неизвестна, механизмы вероятно связаны с нарушением функции Т-
Гипер-IgE-синдром (синдром Джоба)
Причины: частота неизвестна, механизмы вероятно связаны с нарушением функции Т-

Слайд 27Комбинированные иммунодефициты
Комбинированные иммунодефициты

Слайд 28Особенности клинических проявлений вторичных иммунодефицитов при недостаточности клеточного иммунитета
Клинические проявления с первых
Особенности клинических проявлений вторичных иммунодефицитов при недостаточности клеточного иммунитета
Клинические проявления с первых

Слайд 29У взрослых
Первичные комбинированные иммунодефициты и иммунодефициты по клеточному звену практически не встречаются
У взрослых
Первичные комбинированные иммунодефициты и иммунодефициты по клеточному звену практически не встречаются

Слайд 30Синдром Гуда (тимома с наличием иммунодефицита)
Причины: редкий комбинированный В- и Т-клеточный первичный
Синдром Гуда (тимома с наличием иммунодефицита)
Причины: редкий комбинированный В- и Т-клеточный первичный

Слайд 31Синдром Гуда (тимома с наличием иммунодефицита)
Клиника: часто бессимптомно, тимома диагностируется случайно (при
Синдром Гуда (тимома с наличием иммунодефицита)
Клиника: часто бессимптомно, тимома диагностируется случайно (при

Слайд 32Синдром Гуда (тимома с наличием иммунодефицита)
Лечение: оперативное лечение, постоянная заместительная терапия внутривенным
Синдром Гуда (тимома с наличием иммунодефицита)
Лечение: оперативное лечение, постоянная заместительная терапия внутривенным

Слайд 33Недостаточность фагоцитарного звена
Недостаточность фагоцитарного звена

Слайд 34Особенности клинических проявлений при нарушении неспецифического иммунитета
Длительно текущие и рецидивирующие инфекции
Скудность клинических
Особенности клинических проявлений при нарушении неспецифического иммунитета
Длительно текущие и рецидивирующие инфекции
Скудность клинических

Слайд 35Хроническая гранулематозная болезнь (ХГБ)
Причины: имеется дефект внутриклеточного бактериального киллинга (нарушено образование супероксида,
Хроническая гранулематозная болезнь (ХГБ)
Причины: имеется дефект внутриклеточного бактериального киллинга (нарушено образование супероксида,

Слайд 36Циклическая нейтропения
Причина: Циклическое изменение количества нейтрофилов; цикл обычно 21 день
Клиника: язвы околоротовой
Циклическая нейтропения
Причина: Циклическое изменение количества нейтрофилов; цикл обычно 21 день
Клиника: язвы околоротовой

Слайд 37Патология системы комплемента
Патология системы комплемента

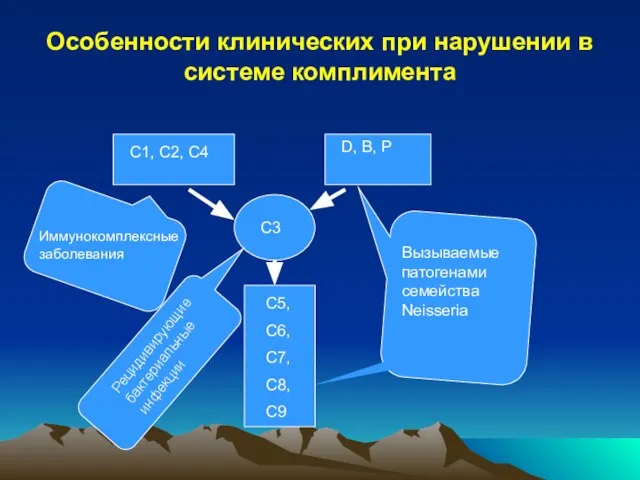
Слайд 38Особенности клинических при нарушении в системе комплимента
C1, C2, C4
D, B, P
C3
C5,
C6,
C7,
C8,
C9
Иммунокомплексные заболевания
Рецидивирующие
Особенности клинических при нарушении в системе комплимента
C1, C2, C4
D, B, P
C3
C5,
C6,
C7,
C8,
C9
Иммунокомплексные заболевания
Рецидивирующие
Слайд 39Наследственный ангионевротический отек
Причины: аутосомно-доминантное тип, характеризующийся снижением активности ингибитора С1-эстеразу, что ведет
Наследственный ангионевротический отек
Причины: аутосомно-доминантное тип, характеризующийся снижением активности ингибитора С1-эстеразу, что ведет

Слайд 40Иммуностимуляторы, применяемые для лечения первичных иимунодефицитов
Иммуноглобулины: иммуноглобулин человека нормальный для в/в введения,
Иммуностимуляторы, применяемые для лечения первичных иимунодефицитов
Иммуноглобулины: иммуноглобулин человека нормальный для в/в введения,

Слайд 41СПАСИБО
СПАСИБО

Слайд 42Клинико-анамнестические признаки иммунодефицита
Частые простудные заболевания (более 4 раз в год)
Рецидивирующий герпес (4-6
Клинико-анамнестические признаки иммунодефицита
Частые простудные заболевания (более 4 раз в год)
Рецидивирующий герпес (4-6

Слайд 43Ключи к диагностике ПЕРВИЧНОГО иммунодефицита
1. Возраст до 5 лет
2. Главные*
2.1.Частые заболевания отитом
Ключи к диагностике ПЕРВИЧНОГО иммунодефицита
1. Возраст до 5 лет
2. Главные*
2.1.Частые заболевания отитом

Слайд 44Ключи к диагностике ВТОРИЧНОГО иммунодефицита
1. В анамнезе
1.1.Вирусные инфекции (HIV, EBV, CMV, вирусы
Ключи к диагностике ВТОРИЧНОГО иммунодефицита
1. В анамнезе
1.1.Вирусные инфекции (HIV, EBV, CMV, вирусы

Слайд 45Ключи к диагностике ВТОРИЧНОГО иммунодефицита
2. Главные клинические признаки
2.1. Частые простудные заболевания (более
Ключи к диагностике ВТОРИЧНОГО иммунодефицита
2. Главные клинические признаки
2.1. Частые простудные заболевания (более

 Способы размножения животных. Оплодотворение
Способы размножения животных. Оплодотворение 129626, г.Москва, Графский переулок д.9, стр.2. тел.:(495)9335900, 3635612(13). Факс:9335901. e-mail:
129626, г.Москва, Графский переулок д.9, стр.2. тел.:(495)9335900, 3635612(13). Факс:9335901. e-mail:  Волшебная сила энергии
Волшебная сила энергии « Древнейший Рим»
« Древнейший Рим» Fabbrica Apparecchiature ElettroMeccaniche e Affini Компания 1 на мировом рынке кофейного оборудования: - 68% импортируется более чем 100 стран, 700 дистрибьюте
Fabbrica Apparecchiature ElettroMeccaniche e Affini Компания 1 на мировом рынке кофейного оборудования: - 68% импортируется более чем 100 стран, 700 дистрибьюте Презентація(1)(1)
Презентація(1)(1) Освещение. Свет и тень
Освещение. Свет и тень Средства обучения
Средства обучения Выставка объединения Умелая иголочка
Выставка объединения Умелая иголочка Арт фестиваль All is Art. Проект
Арт фестиваль All is Art. Проект Реформы М М Сперанского
Реформы М М Сперанского Описание системы работы
Описание системы работы Разработка и презентация проектов ОФО-1
Разработка и презентация проектов ОФО-1 Эстафета олимпийского огня в Перми 2014 г
Эстафета олимпийского огня в Перми 2014 г Презентация-1 (1)
Презентация-1 (1) English language
English language  Проектная и учебно-исследовательская деятельность учащихся
Проектная и учебно-исследовательская деятельность учащихся Живой уголок
Живой уголок Медиапроект
Медиапроект Великие географические путешественники и их открытия
Великие географические путешественники и их открытия Форменные элементы крови
Форменные элементы крови Презентация на тему Архитектура барокко
Презентация на тему Архитектура барокко  Общее понятие контроля
Общее понятие контроля Устройство храма
Устройство храма Психологические законы
Психологические законы Одежда. Европа XII – XIX веков
Одежда. Европа XII – XIX веков Презентация на тему Оман
Презентация на тему Оман  Набережные Челны - город экономического роста
Набережные Челны - город экономического роста