- Популяционная генетика

Содержание
- 2. Исследование полногеномной ассоциации Ищем SNP… связанный с фенотипом. Цель: Объяснять Понимание Механизмы Терапия Предсказывать Вмешательство Профилактика
- 3. Определение Любая связь между двумя измеренными величинами, которая делает их статистически зависимыми. Наследственность Доля дисперсии, объясняемая
- 4. Определение Любая связь между двумя измеренными величинами, которая делает их статистически зависимыми. Наследственность Доля дисперсии, объясняемая
- 5. Почему? Окружающая среда, взаимодействие генов и окружающей среды Сложные черты, небольшие эффекты, редкие варианты Уровни экспрессии
- 6. Кейс-контроль Четко определенный «случай» Известная наследственность Вариации Количественные фенотипические данные Например: Рост, концентрация биомаркеров Явные модели
- 7. Процесс Визуализация Филогенетика PCA Коррекция данных Геномный контроль Регрессия по основным компонентам PCA Стратификация населения II
- 8. Что же там, в наших данных? Применим PCA - Метод главных компонент Для начала рассмотрим простые
- 9. Пример: планеты Солнечной системы Как вы думаете, есть какая корреляция между колонками?
- 10. Пример: планеты Солнечной системы Корреляция между колонками? Три измерения явно излишни
- 11. Что показывает PCA? Сколько компонент оставить? Два подхода Все со стандартными отклонениями больше 1 Совокупная пропорция
- 12. Результаты Расстояние Плотность Диаметр Saturn PC1 PC2
- 13. Эффект нормализации: даем равный шанс разным группам измерений С нормализацией 2 компоненты Без нормализации 1 главная
- 14. Теперь попробуйте сами # загрузите файл с данными планет # planets.csv Planets=read.csv('Planets.csv', row.names = 1); Planets
- 15. Повторим упражнение с данными о странах мира Данные Вопросы: Нужно ли проводить нормализацию? Сколько главных компонент
- 16. Результаты
- 17. Простая карта install.packages(“maptools”) library(maptools) data(wrld_simpl) myCountries = wrld_simpl@data$NAME %in% row.names(Countries) plot(wrld_simpl, col = c(gray(.80), "red")[myCountries+1])
- 18. Не совсем простая карта MAX_INCOME=max(Countries$average.income) # добавили колонку Countries$index=round(10*Countries$average.income/MAX_INCOME); Countries COL=rainbow(10) # задали цвета ALL_COUNTRIES=as.data.frame(cbind( as.character(wrld_simpl@data$NAME
- 19. K-MEANS install.packages(“cluster”) library(cluster) Countries_clusters=kmeans(Countries[,1:3], centers=4, nstart=25) clusplot(Countries, Countries_clusters$cluster,labels=3, color=TRUE) ccs=data.frame(sapply(Countries[,1:3], scale)) ##нормализация rownames(ccs)=rownames(Countries) Countries_clusters_scaled=kmeans(ccs, centers=4, nstart=25)
- 22. Возвращаемся к нуклеотидам Скачайте данные в вашу рабочую директорию sativas413.ped sativas413.fam sativas413.map sativas413.pheno sativas413.csv Мастерская GWAS
- 23. Библиотеки #install packages install.packages(c("poolr","qqman","BGLR","rrBLUP","DT", "dplyr")) install.packages(c("rnaturalearth",'rnaturalearthdata','rgeos','ggspatial')) devtools::install_github("dkahle/ggmap", ref = "tidyup") if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
- 24. Библиотеки library(rrBLUP) library(BGLR) library(DT) library(SNPRelate) library(dplyr) library(qqman) library(poolr) library(OpenStreetMap) library(rjson) library(rgdal) library(RgoogleMaps) library(mapproj) library(sf) library(OpenStreetMap) library(ggplot2)
- 25. Шаг 1. Подготовка данных SNP в R rm (список = ls ()) setwd («# ваш рабочий
- 26. Шаг 2. Считайте данные фенотипа в R ## прочитать фенотип ris.pheno header = TRUE, stringsAsFactors =
- 27. Шаг 3. Фильтрация данных SNP # Здесь мы будем использовать цикл for, чтобы определить недостающие и
- 28. Шаг 4. Структура популяции # Создать файл геноматрицы и назначить имена строк и столбцов из файлов
- 29. Шаг 5. PCA #PCA анализ pca # график результатов PCA pca plot(pca $ EV2, pca $
- 30. Шаг 5а. Визуализация PCA # Извлечь информацию о местоположении и добавить выходной файл PCA pca_country as.numeric(pca$EV4),as.numeric(pca_2$Latitude),
- 31. Анализ PCA: цвет по населению и по географии Мастерская GWAS
- 33. Аллели в отдельных локусах зависимы друг от друга Проблема? Да и нет Слишком много LD -
- 34. Стандартный GWAS Одномерные методы Использования взаимодействий Многовариантные методы Методы штрафной регрессии (LASSO) Факториальные методы (ФС на
- 35. Вариации Тестирование Точный критерий Фишера, критерий тенденции Кохрана-Армитиджа, критерий хи-квадрат, дисперсионный анализ Золотой стандарт - точный
- 36. Сильные стороны Недостатки Простота Вычислительно быстро Консервативный Легко интерпретировать Многомерная система, одномерная структура Размер эффекта отдельных
- 37. А как насчет взаимодействия? Тестирование на ассоциацию
- 38. Многовариантные методы Тестирование на ассоциацию Штрафная регрессия LASSO Регрессия хребта (ридж Регрессия) Нейронные сети Штрафная регрессия
- 39. GWAS Ридж регрессия (Метод регуляризации Тихонова) с использованием пакета rrBLUP Ридж регрессия BLUP (Meuwissen et al.
- 40. Код R: установите и загрузите необходимые библиотеки install.packages (c («poolr», «qqman», «BGLR», «rrBLUP», «DT», «SNPRelate», «dplyr»,
- 41. GWAS # создаем файл geno для анализа GWAS пакета rrBLUP geno_final dim(Geno1) # создаем фенофайл pheno_final
- 42. Мастерская GWAS
- 43. Использовать поиск по SNP https://snp-seek.irri.org/ Мастерская GWAS
- 44. Есть много новых инструментов Однако, если вы освоите старый добрый rrBLUP, это поможет понять другие. Теперь
- 45. Подход к прогнозированию географической структуры населения (GPS) Сделать вывод о происхождении человека из полногеномной коллекции маркеров,
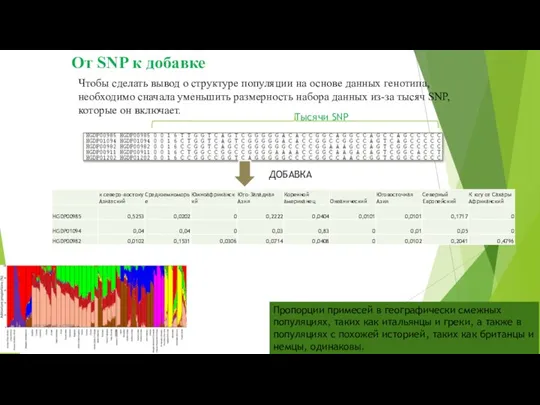
- 46. Чтобы сделать вывод о структуре популяции на основе данных генотипа, необходимо сначала уменьшить размерность набора данных
- 47. Мастерская GWAS
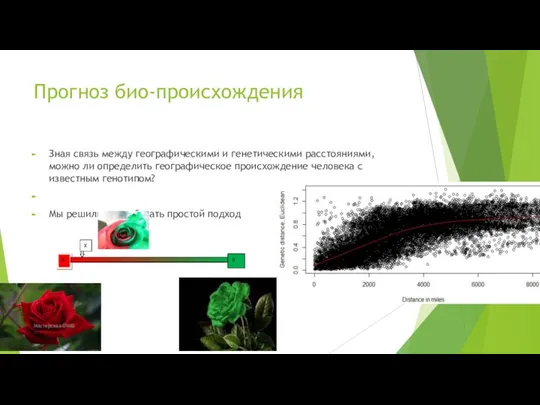
- 48. Прогноз био-происхождения Зная связь между географическими и генетическими расстояниями, можно ли определить географическое происхождение человека с
- 49. Неизвестные образцы Мастерская GWAS
- 50. GPS точно назначен ~ 100% всех людей в свои континентальные регионы 80% всех людей в страну
- 51. Моделирующая добавка
- 52. 1001 Genomes - Каталог генетической изменчивости Arabidopsis thaliana. Образцы из 22 стран Массив SNP генотипа 250К
- 53. Структура населения Мэтью В. Хортон и др. Полногеномные паттерны генетической изменчивости во всем мире образцов A.
- 54. Фильтрация SNP Мы взяли файлы вариантов, доступные на http://1001genomes.org/data/MPI/MPICWang2013/releases/current/. отфильтрованы инделы и неаутосомные варианты. Обнаружено около
- 55. ДОБАВКА Мы выполнили несколько анализов ADMIXTURE с числом предковых популяций K от 3 до 20, а
- 56. Примесь Мастерская GWAS
- 57. Проверка GPS по одному разу Процент популяций, которые точно нанесены на карту 60% Среднее расстояние до
- 58. Исходная гипотеза Географическое положение Arabidopsis должно быть связано с холодоустойчивостью. Более холодный климат в Северном полушарии
- 59. GPS-анализ O. sativa Мастерская GWAS
- 60. Точность для риса Среднее расстояние: 4043 км Мастерская GWAS
- 61. Прогнозирование GPS после SNP и географической фильтрации Среднее расстояние: 1141 км Мастерская GWAS
- 62. Почему не работает с рисом? Рис не может выбрать свою вторую половинку, а арабидопсис может. Мастерская
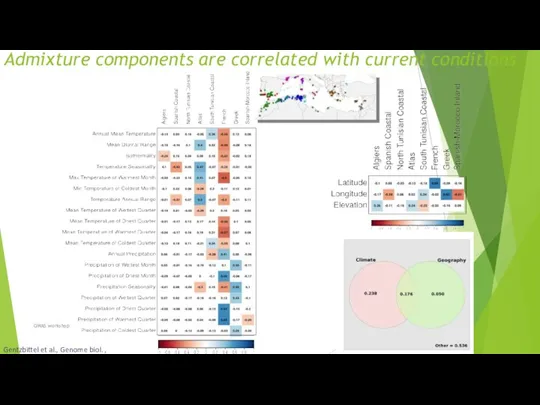
- 63. Наборы данных: WorldClime и SoilDB Компоненты примеси больше зависят от климата, чем от параметров почвы. Климат
- 64. Часть 1 заключение Мы умеем моделировать дикие виды - не так уж и много с одомашненными
- 65. Семейство бобовых (бобовых) растений Возможность установления клубенькового симбиоза Высокая синтения к бобовым культурам Небольшой диплоидный геном
- 66. Проект NSF HapMap Текущее состояние: 288 последовательных образцов. Итого: 16.516.721 SNP 30 строк при> 20X Остальные
- 67. 12.11.2020 Зерновые бобовые Кормовые бобовые Люцерна Клевер Соя Нута Фасоль Высокая сельскохозяйственная ценность Горох Арахис Чечевица
- 68. Данные HapMap для M. truncatula Текущее состояние: 288 последовательных образцов. # SNP Chr1: 1,508,346 Chr2: 1.964.419
- 69. M. truncatula спонтанно встречается по всему Средиземноморскому бассейну. Доступно несколько коллекций: DZ, TN, FR, AU, US,….
- 70. Некоторые наследственные геномы могут быть адаптированы к местным биоклиматическим переменным. Мы искали предполагаемую связь с 19
- 71. PCA определяет две субпопуляции Medicago среди 262 образцов. 40 000 случайно выбранных SNP В соответствии с
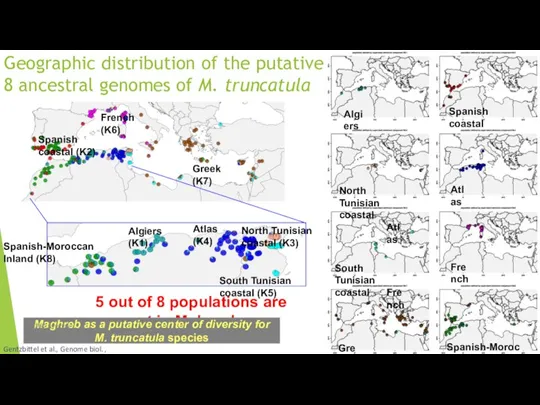
- 72. Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS Medicago, вероятно, будет иметь 8
- 73. Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS Gentzbittel et al., Genome biol.
- 74. Предсказано местонахождение 17 неизвестных образцов, включая эталонный геном Jemalong-A17. Точность процедуры «Leave-one Out» Линейная связь между
- 75. Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS Мастерская GWAS
- 76. Geographic distribution of the putative 8 ancestral genomes of M. truncatula Spanish coastal Algiers North Tunisian
- 77. Last glaciations & main glacial refugia may explain M. truncatula population structure -20.000 BP GWAS workshop
- 78. Admixture components are correlated with current conditions Gentzbittel et al., Genome biol. , 2019 GWAS workshop
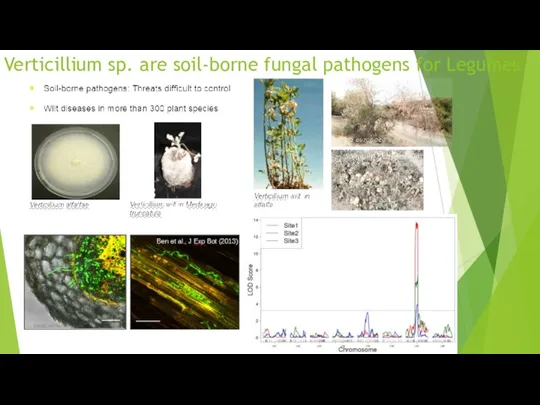
- 79. Verticillium sp. are soil-borne fungal pathogens for Legumes GWAS workshop
- 80. Ancestral genomes may present different levels of resistance to Verticillium wilt V. alfalfae symptoms on susceptible
- 81. Ancestral genomes present different levels of resistance to Verticillium wilt Linear Model (242 Mt HAPMAP accessions)
- 82. Can phenotype be predicted using WhoGem? Use plant model (Medicago truncatula) to test applicability of admixture
- 83. Experimental validation of predicted resistance levels 21/29 samples that are predicted to be resistant were resistant
- 84. WhoGem models can be significant predictors of quantitative functional traits in plants Comparisons of accuracy of
- 85. What is missing? Distance between two points should not be just geometric distance. Add: Mode of
- 86. GWAS workshop
- 87. GWAS workshop
- 88. GWAS workshop
- 89. This methodology may serve as a basis for analyses in other plant species and for other
- 90. Practice Admixture.csv Pheno.csv GWAS workshop
- 91. ADM=read.csv("Admixture.csv",row.names = 1) # Instruction to name bars: barNaming retVec for (k in 2:length(vec)) { if
- 92. Clustered vectors GWAS workshop
- 93. Merge datasets ADM=read.csv('Admixture.csv',row.names = 1); TEST=subset(ADM, is.na(ADM$Lat)) REF=subset(ADM, !is.na(ADM$Lat)); Phen=read.csv('pheno.csv',row.names = 1); head(Phen) #merge reference adm
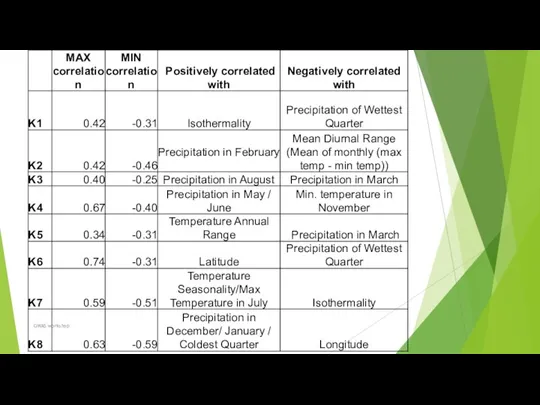
- 94. DATA=AMDPH[,c(4:6,8:15,17)]; cor(DATA) GWAS workshop
- 95. ##GPS M=25; for(i in 1:dim(TEST)[1]){ Y=TEST[i,7:14];Y DIST=c() for(j in 1:dim(AMDPH)[1]){ Z=AMDPH[j,8:15];Z d=sum((Y-Z)^2);d DIST=c(DIST,d) } I=order(DIST)[1:M];I Ph=AMDPH$MaxSymptomScore[I];Ph
- 96. Results GWAS workshop
- 97. #GLM library(lmtest); library(dplyr); library(car); library(rcompanion) #set up formulas formula=formula(MaxSymptomScore~K1+K2+K3+K4+K5+K6+K7+K8) formula0=MaxSymptomScore ~ 1 #initialize models model.null =
- 98. Laurent Gentzbittel, Ecolab, Toulouse, France/Skoltech, Moscow, Russia Nevin Young's lab, Univ. of Minnesota, USA Sergey Nuzdhin's
- 100. Скачать презентацию
Слайд 2Исследование полногеномной ассоциации
Ищем SNP…
связанный с фенотипом.
Цель:
Объяснять
Понимание
Механизмы
Терапия
Предсказывать
Вмешательство
Профилактика
Так что же такое GWAS?
Исследование полногеномной ассоциации
Ищем SNP…
связанный с фенотипом.
Цель:
Объяснять
Понимание
Механизмы
Терапия
Предсказывать
Вмешательство
Профилактика
Так что же такое GWAS?

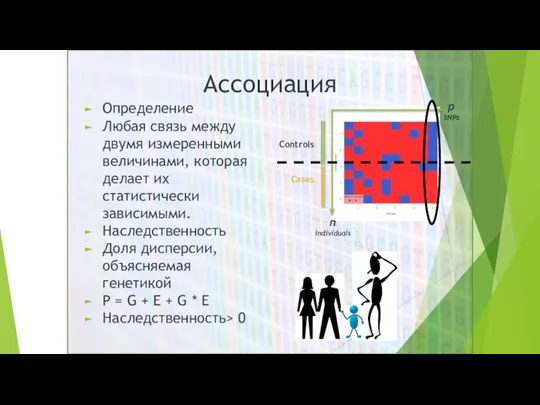
Слайд 3Определение
Любая связь между двумя измеренными величинами, которая делает их статистически зависимыми.
Наследственность
Доля дисперсии,
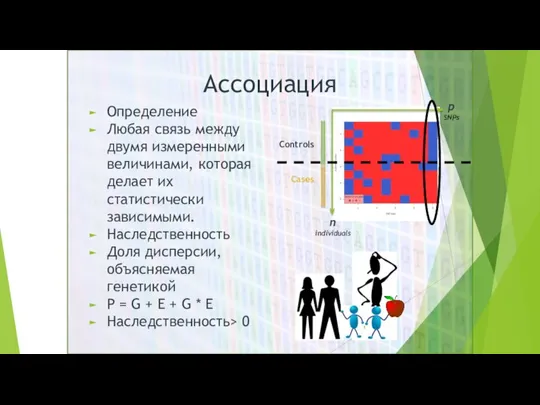
Определение
Любая связь между двумя измеренными величинами, которая делает их статистически зависимыми.
Наследственность
Доля дисперсии,
Слайд 4Определение
Любая связь между двумя измеренными величинами, которая делает их статистически зависимыми.
Наследственность
Доля дисперсии,
Определение
Любая связь между двумя измеренными величинами, которая делает их статистически зависимыми.
Наследственность
Доля дисперсии,
Слайд 5Почему?
Окружающая среда, взаимодействие генов и окружающей среды
Сложные черты, небольшие эффекты, редкие варианты
Уровни
Почему?
Окружающая среда, взаимодействие генов и окружающей среды
Сложные черты, небольшие эффекты, редкие варианты
Уровни

Слайд 6Кейс-контроль
Четко определенный «случай»
Известная наследственность
Вариации
Количественные фенотипические данные
Например: Рост, концентрация биомаркеров
Явные модели
Например. Доминантный или
Кейс-контроль
Четко определенный «случай»
Известная наследственность
Вариации
Количественные фенотипические данные
Например: Рост, концентрация биомаркеров
Явные модели
Например. Доминантный или

Слайд 7Процесс
Визуализация
Филогенетика
PCA
Коррекция данных
Геномный контроль
Регрессия по основным компонентам PCA
Стратификация населения II
Процесс
Визуализация
Филогенетика
PCA
Коррекция данных
Геномный контроль
Регрессия по основным компонентам PCA
Стратификация населения II

Слайд 8Что же там, в наших данных? Применим PCA -
Метод главных компонент
Для
Что же там, в наших данных? Применим PCA -
Метод главных компонент
Для

Слайд 9Пример: планеты Солнечной системы
Как вы думаете, есть какая корреляция между колонками?
Пример: планеты Солнечной системы
Как вы думаете, есть какая корреляция между колонками?

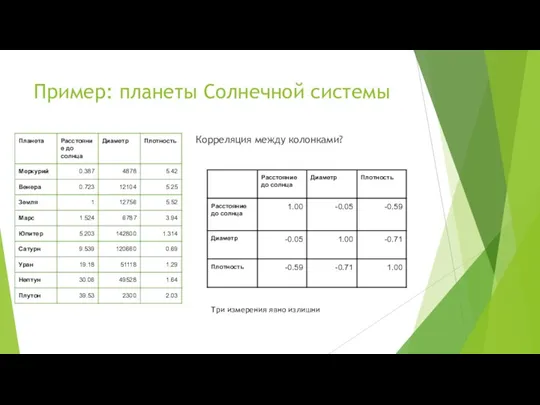
Слайд 10Пример: планеты Солнечной системы
Корреляция между колонками?
Три измерения явно излишни
Пример: планеты Солнечной системы
Корреляция между колонками?
Три измерения явно излишни
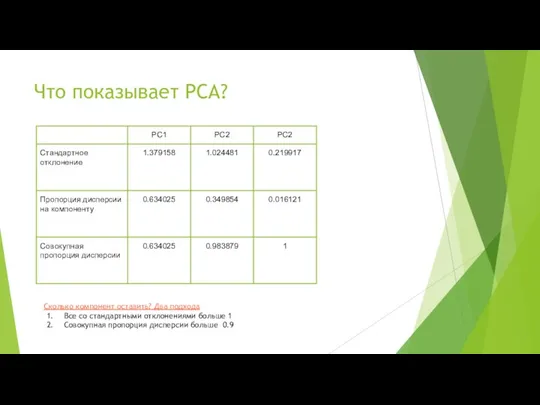
Слайд 11Что показывает PCA?
Сколько компонент оставить? Два подхода
Все со стандартными отклонениями больше 1
Совокупная
Что показывает PCA?
Сколько компонент оставить? Два подхода
Все со стандартными отклонениями больше 1
Совокупная
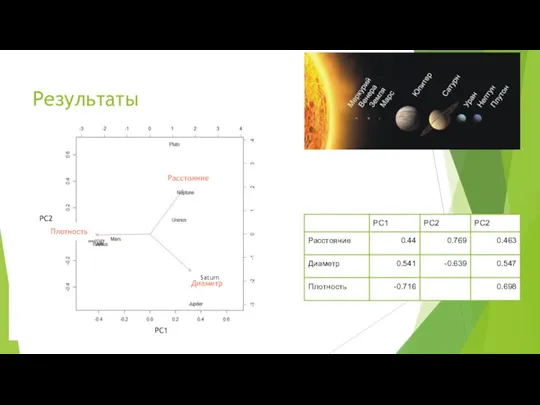
Слайд 12Результаты
Расстояние
Плотность
Диаметр
Saturn
PC1
PC2
Результаты
Расстояние
Плотность
Диаметр
Saturn
PC1
PC2
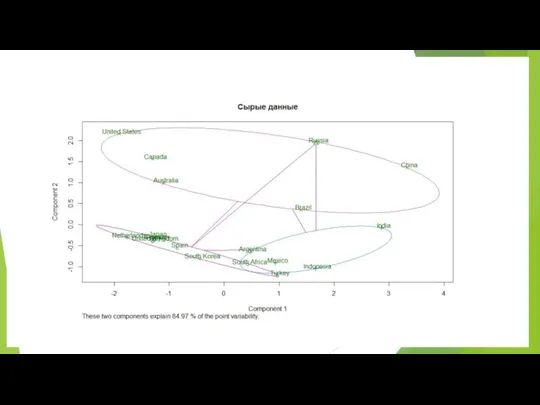
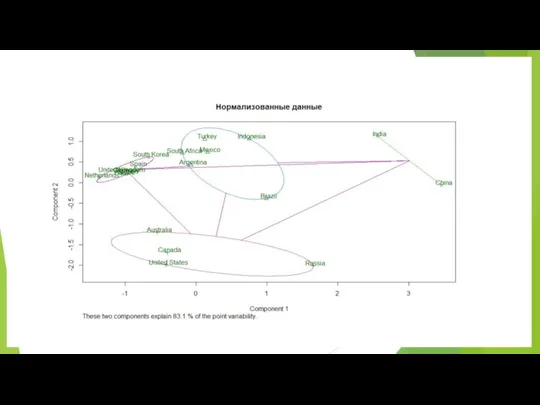
Слайд 13Эффект нормализации: даем равный шанс разным группам измерений
С нормализацией 2 компоненты
Без
Эффект нормализации: даем равный шанс разным группам измерений
С нормализацией 2 компоненты
Без

Слайд 14Теперь попробуйте сами
# загрузите файл с данными планет
# planets.csv
Planets=read.csv('Planets.csv', row.names =
Теперь попробуйте сами
# загрузите файл с данными планет
# planets.csv
Planets=read.csv('Planets.csv', row.names =

Слайд 15Повторим упражнение с данными о странах мира
Данные
Вопросы:
Нужно ли проводить нормализацию?
Сколько главных компонент
Повторим упражнение с данными о странах мира
Данные
Вопросы:
Нужно ли проводить нормализацию?
Сколько главных компонент

Слайд 16Результаты
Результаты

Слайд 17Простая карта
install.packages(“maptools”)
library(maptools)
data(wrld_simpl)
myCountries = wrld_simpl@data$NAME %in% row.names(Countries)
plot(wrld_simpl, col = c(gray(.80), "red")[myCountries+1])
Простая карта
install.packages(“maptools”)
library(maptools)
data(wrld_simpl)
myCountries = wrld_simpl@data$NAME %in% row.names(Countries)
plot(wrld_simpl, col = c(gray(.80), "red")[myCountries+1])
![Простая карта install.packages(“maptools”) library(maptools) data(wrld_simpl) myCountries = wrld_simpl@data$NAME %in% row.names(Countries) plot(wrld_simpl, col = c(gray(.80), "red")[myCountries+1])](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1062822/slide-16.jpg)
Слайд 18Не совсем простая карта
MAX_INCOME=max(Countries$average.income)
# добавили колонку
Countries$index=round(10*Countries$average.income/MAX_INCOME); Countries
COL=rainbow(10) # задали цвета
ALL_COUNTRIES=as.data.frame(cbind( as.character(wrld_simpl@data$NAME ),
Не совсем простая карта
MAX_INCOME=max(Countries$average.income)
# добавили колонку
Countries$index=round(10*Countries$average.income/MAX_INCOME); Countries
COL=rainbow(10) # задали цвета
ALL_COUNTRIES=as.data.frame(cbind( as.character(wrld_simpl@data$NAME ),

Слайд 19K-MEANS
install.packages(“cluster”)
library(cluster)
Countries_clusters=kmeans(Countries[,1:3], centers=4, nstart=25)
clusplot(Countries, Countries_clusters$cluster,labels=3, color=TRUE)
ccs=data.frame(sapply(Countries[,1:3], scale)) ##нормализация
rownames(ccs)=rownames(Countries)
Countries_clusters_scaled=kmeans(ccs, centers=4, nstart=25)
clusplot(ccs, Countries_clusters_scaled$cluster,labels=3, color=TRUE)
K-MEANS
install.packages(“cluster”)
library(cluster)
Countries_clusters=kmeans(Countries[,1:3], centers=4, nstart=25)
clusplot(Countries, Countries_clusters$cluster,labels=3, color=TRUE)
ccs=data.frame(sapply(Countries[,1:3], scale)) ##нормализация
rownames(ccs)=rownames(Countries)
Countries_clusters_scaled=kmeans(ccs, centers=4, nstart=25)
clusplot(ccs, Countries_clusters_scaled$cluster,labels=3, color=TRUE)
![K-MEANS install.packages(“cluster”) library(cluster) Countries_clusters=kmeans(Countries[,1:3], centers=4, nstart=25) clusplot(Countries, Countries_clusters$cluster,labels=3, color=TRUE) ccs=data.frame(sapply(Countries[,1:3], scale)) ##нормализация](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1062822/slide-18.jpg)
Слайд 22Возвращаемся к нуклеотидам
Скачайте данные в вашу рабочую директорию
sativas413.ped
sativas413.fam
sativas413.map
sativas413.pheno
sativas413.csv
Мастерская GWAS
Возвращаемся к нуклеотидам
Скачайте данные в вашу рабочую директорию
sativas413.ped
sativas413.fam
sativas413.map
sativas413.pheno
sativas413.csv
Мастерская GWAS

Слайд 23Библиотеки
#install packages
install.packages(c("poolr","qqman","BGLR","rrBLUP","DT", "dplyr"))
install.packages(c("rnaturalearth",'rnaturalearthdata','rgeos','ggspatial'))
devtools::install_github("dkahle/ggmap", ref = "tidyup")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("SNPRelate")
Библиотеки
#install packages
install.packages(c("poolr","qqman","BGLR","rrBLUP","DT", "dplyr"))
install.packages(c("rnaturalearth",'rnaturalearthdata','rgeos','ggspatial'))
devtools::install_github("dkahle/ggmap", ref = "tidyup")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("SNPRelate")

Слайд 24Библиотеки
library(rrBLUP)
library(BGLR)
library(DT)
library(SNPRelate)
library(dplyr)
library(qqman)
library(poolr)
library(OpenStreetMap)
library(rjson)
library(rgdal)
library(RgoogleMaps)
library(mapproj)
library(sf)
library(OpenStreetMap)
library(ggplot2)
library(sf)
library(rnaturalearth)
library(rnaturalearthdata)
Библиотеки
library(rrBLUP)
library(BGLR)
library(DT)
library(SNPRelate)
library(dplyr)
library(qqman)
library(poolr)
library(OpenStreetMap)
library(rjson)
library(rgdal)
library(RgoogleMaps)
library(mapproj)
library(sf)
library(OpenStreetMap)
library(ggplot2)
library(sf)
library(rnaturalearth)
library(rnaturalearthdata)

Слайд 25Шаг 1. Подготовка данных SNP в R
rm (список = ls ())
setwd («#
Шаг 1. Подготовка данных SNP в R
rm (список = ls ())
setwd («#

Слайд 26Шаг 2. Считайте данные фенотипа в R
## прочитать фенотип
ris.pheno <- read.table ("sativas413.pheno",
Шаг 2. Считайте данные фенотипа в R
## прочитать фенотип
ris.pheno <- read.table ("sativas413.pheno",

Слайд 27Шаг 3. Фильтрация данных SNP
# Здесь мы будем использовать цикл for, чтобы
Шаг 3. Фильтрация данных SNP
# Здесь мы будем использовать цикл for, чтобы

Слайд 28Шаг 4. Структура популяции
# Создать файл геноматрицы и назначить имена строк и
Шаг 4. Структура популяции
# Создать файл геноматрицы и назначить имена строк и

Слайд 29Шаг 5. PCA
#PCA анализ
pca <- snpgdsPCA (geno_44k, snp.id = colnames (Geno1))
# график
Шаг 5. PCA
#PCA анализ
pca <- snpgdsPCA (geno_44k, snp.id = colnames (Geno1))
# график

Слайд 30Шаг 5а. Визуализация PCA
# Извлечь информацию о местоположении и добавить выходной файл
Шаг 5а. Визуализация PCA
# Извлечь информацию о местоположении и добавить выходной файл

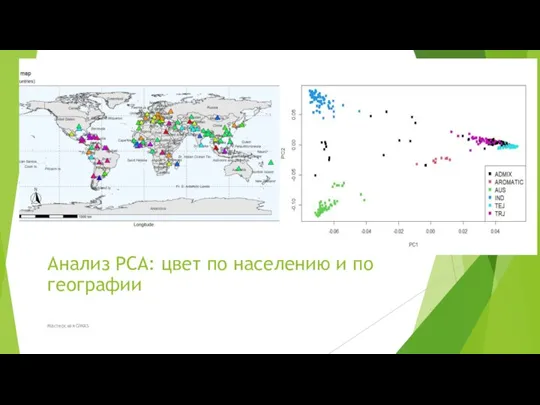
Слайд 31Анализ PCA: цвет по населению и по географии
Мастерская GWAS
Анализ PCA: цвет по населению и по географии
Мастерская GWAS

Слайд 33Аллели в отдельных локусах зависимы друг от друга
Проблема? Да и нет
Слишком много
Аллели в отдельных локусах зависимы друг от друга
Проблема? Да и нет
Слишком много

Слайд 34Стандартный GWAS
Одномерные методы
Использования взаимодействий
Многовариантные методы
Методы штрафной регрессии (LASSO)
Факториальные методы (ФС на основе
Стандартный GWAS
Одномерные методы
Использования взаимодействий
Многовариантные методы
Методы штрафной регрессии (LASSO)
Факториальные методы (ФС на основе

Слайд 35Вариации
Тестирование
Точный критерий Фишера, критерий тенденции Кохрана-Армитиджа, критерий хи-квадрат, дисперсионный анализ
Золотой стандарт -
Тестирование
Точный критерий Фишера, критерий тенденции Кохрана-Армитиджа, критерий хи-квадрат, дисперсионный анализ
Золотой стандарт -

Слайд 36Сильные стороны
Недостатки
Простота
Вычислительно быстро
Консервативный
Легко интерпретировать
Многомерная система, одномерная структура
Размер эффекта отдельных SNP может быть
Сильные стороны
Недостатки
Простота
Вычислительно быстро
Консервативный
Легко интерпретировать
Многомерная система, одномерная структура
Размер эффекта отдельных SNP может быть

Слайд 37А как насчет взаимодействия?
Тестирование на ассоциацию
А как насчет взаимодействия?
Тестирование на ассоциацию
Слайд 38Многовариантные методы
Тестирование на ассоциацию
Штрафная регрессия LASSO
Регрессия хребта (ридж Регрессия)
Нейронные сети
Штрафная регрессия
Байесовские подходы
Факторные
Многовариантные методы
Тестирование на ассоциацию
Штрафная регрессия LASSO
Регрессия хребта (ридж Регрессия)
Нейронные сети
Штрафная регрессия
Байесовские подходы
Факторные

Слайд 39GWAS
Ридж регрессия (Метод регуляризации Тихонова) с использованием пакета rrBLUP
Ридж регрессия BLUP (Meuwissen
GWAS
Ридж регрессия (Метод регуляризации Тихонова) с использованием пакета rrBLUP
Ридж регрессия BLUP (Meuwissen

Слайд 40Код R: установите и загрузите необходимые библиотеки
install.packages (c («poolr», «qqman», «BGLR», «rrBLUP»,
Код R: установите и загрузите необходимые библиотеки
install.packages (c («poolr», «qqman», «BGLR», «rrBLUP»,

Слайд 41GWAS
# создаем файл geno для анализа GWAS пакета rrBLUP
geno_final <- data.frame (marker
GWAS
# создаем файл geno для анализа GWAS пакета rrBLUP
geno_final <- data.frame (marker

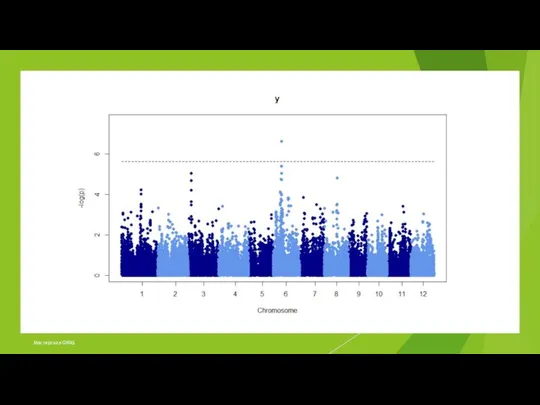
Слайд 42Мастерская GWAS
Мастерская GWAS
Слайд 43Использовать поиск по SNP
https://snp-seek.irri.org/
Мастерская GWAS
Использовать поиск по SNP
https://snp-seek.irri.org/
Мастерская GWAS

Слайд 44Есть много новых инструментов
Однако, если вы освоите старый добрый rrBLUP, это поможет
Есть много новых инструментов
Однако, если вы освоите старый добрый rrBLUP, это поможет

Слайд 45Подход к прогнозированию географической структуры населения (GPS)
Сделать вывод о происхождении человека из
Подход к прогнозированию географической структуры населения (GPS)
Сделать вывод о происхождении человека из

Слайд 46Чтобы сделать вывод о структуре популяции на основе данных генотипа, необходимо сначала
Чтобы сделать вывод о структуре популяции на основе данных генотипа, необходимо сначала
Слайд 47Мастерская GWAS
Мастерская GWAS

Слайд 48Прогноз био-происхождения
Зная связь между географическими и генетическими расстояниями, можно ли определить географическое
Прогноз био-происхождения
Зная связь между географическими и генетическими расстояниями, можно ли определить географическое
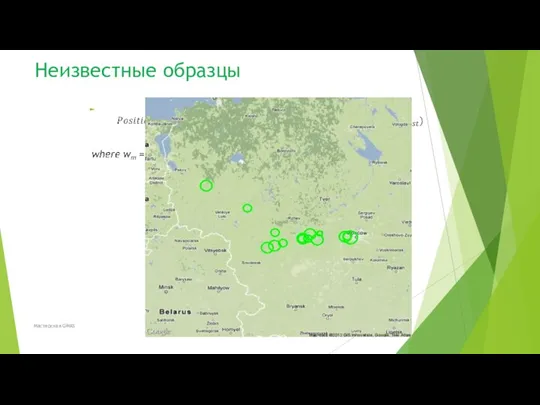
Слайд 49Неизвестные образцы
Мастерская GWAS
Неизвестные образцы
Мастерская GWAS
Слайд 50GPS точно назначен
~ 100% всех людей в свои континентальные регионы
80%
GPS точно назначен
~ 100% всех людей в свои континентальные регионы
80%

Слайд 51Моделирующая добавка
Моделирующая добавка

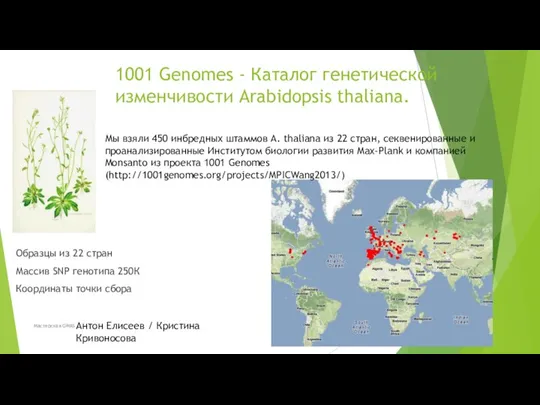
Слайд 521001 Genomes - Каталог генетической изменчивости Arabidopsis thaliana.
Образцы из 22 стран
Массив SNP
1001 Genomes - Каталог генетической изменчивости Arabidopsis thaliana.
Образцы из 22 стран
Массив SNP
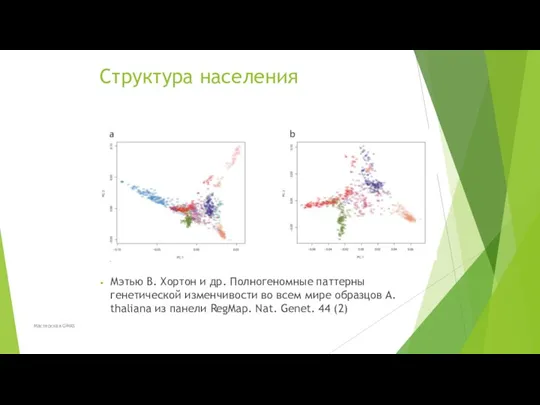
Слайд 53Структура населения
Мэтью В. Хортон и др. Полногеномные паттерны генетической изменчивости во всем
Структура населения
Мэтью В. Хортон и др. Полногеномные паттерны генетической изменчивости во всем
Слайд 54Фильтрация SNP
Мы взяли файлы вариантов, доступные на http://1001genomes.org/data/MPI/MPICWang2013/releases/current/.
отфильтрованы инделы и неаутосомные варианты.
Фильтрация SNP
Мы взяли файлы вариантов, доступные на http://1001genomes.org/data/MPI/MPICWang2013/releases/current/.
отфильтрованы инделы и неаутосомные варианты.

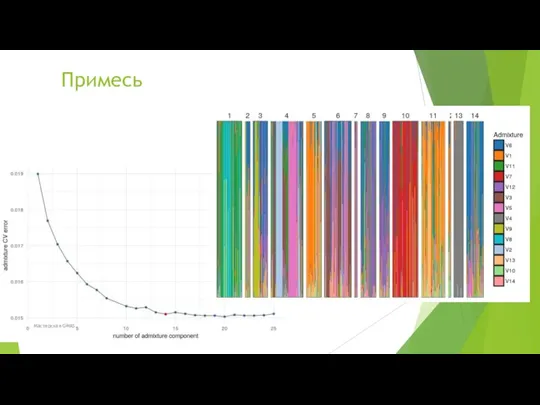
Слайд 55ДОБАВКА
Мы выполнили несколько анализов ADMIXTURE с числом предковых популяций K от 3
ДОБАВКА
Мы выполнили несколько анализов ADMIXTURE с числом предковых популяций K от 3

Слайд 56Примесь
Мастерская GWAS
Примесь
Мастерская GWAS
Слайд 57Проверка GPS по одному разу
Процент популяций, которые точно нанесены на карту 60%
Среднее
Проверка GPS по одному разу
Процент популяций, которые точно нанесены на карту 60%
Среднее

Слайд 58Исходная гипотеза
Географическое положение Arabidopsis должно быть связано с холодоустойчивостью. Более холодный климат
Исходная гипотеза
Географическое положение Arabidopsis должно быть связано с холодоустойчивостью. Более холодный климат

Слайд 59GPS-анализ O. sativa
Мастерская GWAS
GPS-анализ O. sativa
Мастерская GWAS

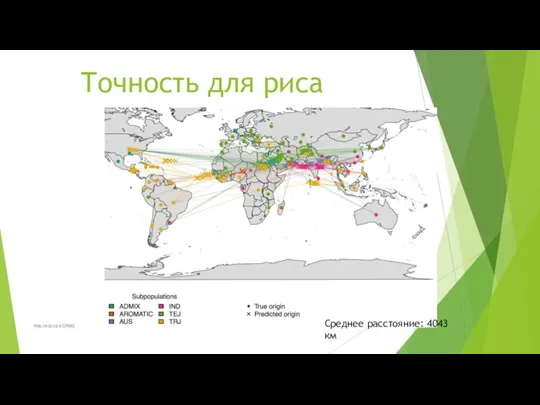
Слайд 60Точность для риса
Среднее расстояние: 4043 км
Мастерская GWAS
Точность для риса
Среднее расстояние: 4043 км
Мастерская GWAS
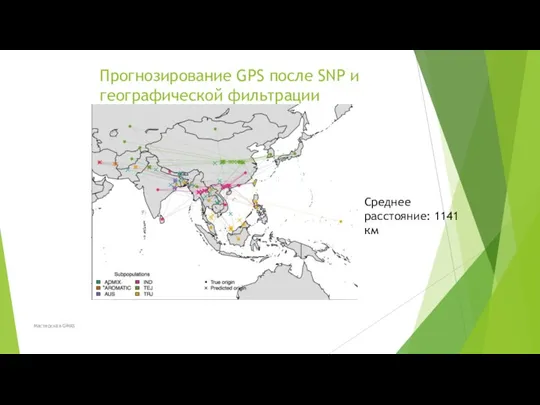
Слайд 61Прогнозирование GPS после SNP и географической фильтрации
Среднее расстояние: 1141 км
Мастерская GWAS
Прогнозирование GPS после SNP и географической фильтрации
Среднее расстояние: 1141 км
Мастерская GWAS
Слайд 62Почему не работает с рисом?
Рис не может выбрать свою вторую половинку, а
Почему не работает с рисом?
Рис не может выбрать свою вторую половинку, а

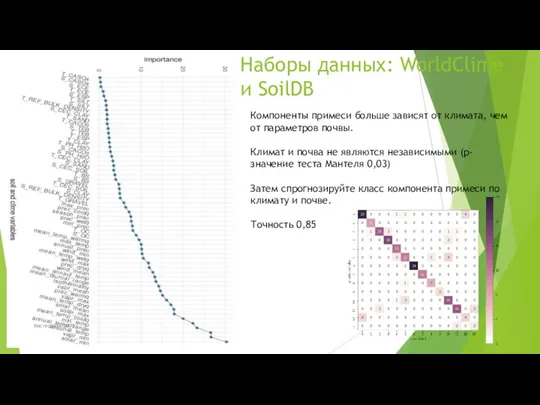
Слайд 63Наборы данных: WorldClime и SoilDB
Компоненты примеси больше зависят от климата, чем от
Наборы данных: WorldClime и SoilDB
Компоненты примеси больше зависят от климата, чем от
Слайд 64Часть 1 заключение
Мы умеем моделировать дикие виды - не так уж и
Часть 1 заключение
Мы умеем моделировать дикие виды - не так уж и

Слайд 65Семейство бобовых (бобовых) растений
Возможность установления клубенькового симбиоза
Высокая синтения к бобовым культурам
Небольшой диплоидный
Семейство бобовых (бобовых) растений
Возможность установления клубенькового симбиоза
Высокая синтения к бобовым культурам
Небольшой диплоидный

Слайд 66Проект NSF HapMap
Текущее состояние: 288 последовательных образцов.
Итого: 16.516.721 SNP
30 строк при> 20X
Остальные
Проект NSF HapMap
Текущее состояние: 288 последовательных образцов.
Итого: 16.516.721 SNP
30 строк при> 20X
Остальные

Слайд 6712.11.2020
Зерновые бобовые
Кормовые бобовые
Люцерна
Клевер
Соя
Нута
Фасоль
Высокая сельскохозяйственная ценность
Горох
Арахис
12.11.2020
Зерновые бобовые
Кормовые бобовые
Люцерна
Клевер
Соя
Нута
Фасоль
Высокая сельскохозяйственная ценность
Горох
Арахис

Слайд 68Данные HapMap для M. truncatula
Текущее состояние: 288 последовательных образцов.
# SNP
Chr1:
Данные HapMap для M. truncatula
Текущее состояние: 288 последовательных образцов.
# SNP
Chr1:

Слайд 69M. truncatula спонтанно встречается по всему Средиземноморскому бассейну.
Доступно несколько коллекций: DZ, TN,
M. truncatula спонтанно встречается по всему Средиземноморскому бассейну.
Доступно несколько коллекций: DZ, TN,

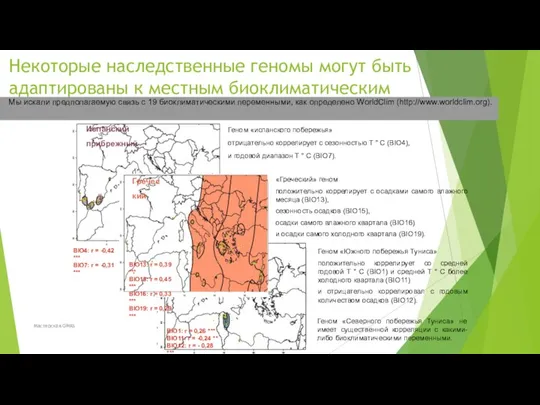
Слайд 70Некоторые наследственные геномы могут быть адаптированы к местным биоклиматическим переменным.
Мы искали предполагаемую
Некоторые наследственные геномы могут быть адаптированы к местным биоклиматическим переменным.
Мы искали предполагаемую
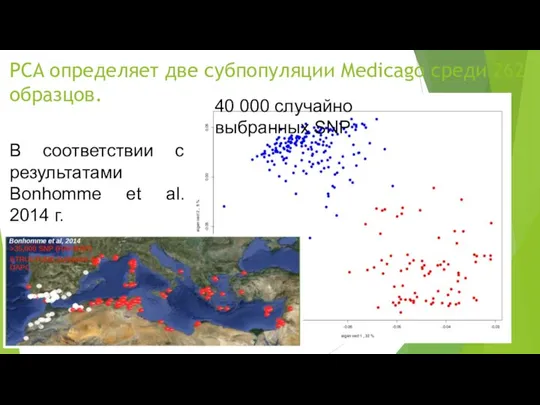
Слайд 71PCA определяет две субпопуляции Medicago среди 262 образцов.
40 000 случайно выбранных SNP
В
PCA определяет две субпопуляции Medicago среди 262 образцов.
40 000 случайно выбранных SNP
В
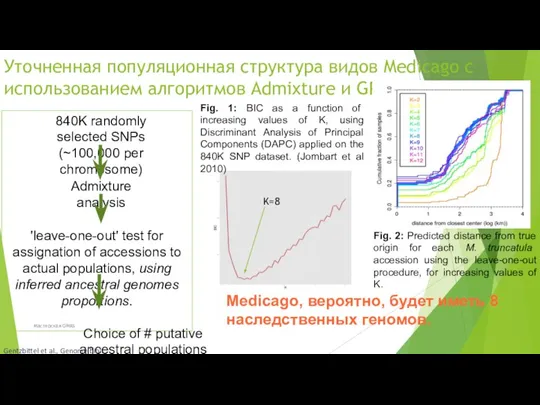
Слайд 72Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS
Medicago, вероятно,
Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS
Medicago, вероятно,
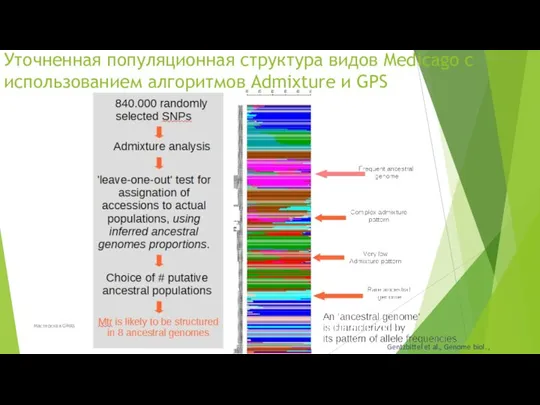
Слайд 73Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS
Gentzbittel et
Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS
Gentzbittel et
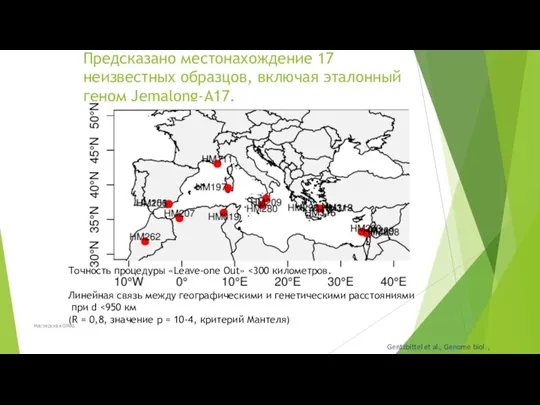
Слайд 74Предсказано местонахождение 17 неизвестных образцов, включая эталонный геном Jemalong-A17.
Точность процедуры «Leave-one Out»
Предсказано местонахождение 17 неизвестных образцов, включая эталонный геном Jemalong-A17.
Точность процедуры «Leave-one Out»
Слайд 75Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS
Мастерская GWAS
Уточненная популяционная структура видов Medicago с использованием алгоритмов Admixture и GPS
Мастерская GWAS

Слайд 76Geographic distribution of the putative 8 ancestral genomes of M. truncatula
Spanish coastal
Algiers
North
Geographic distribution of the putative 8 ancestral genomes of M. truncatula
Spanish coastal
Algiers
North
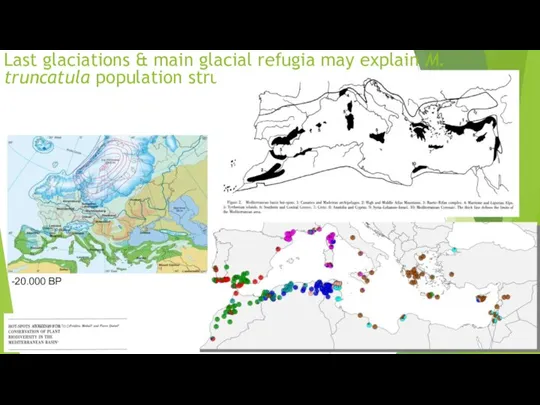
Слайд 77Last glaciations & main glacial refugia may explain M. truncatula population structure
-20.000
Last glaciations & main glacial refugia may explain M. truncatula population structure
-20.000
Слайд 78Admixture components are correlated with current conditions
Gentzbittel et al., Genome biol. ,
Admixture components are correlated with current conditions
Gentzbittel et al., Genome biol. ,
Слайд 79Verticillium sp. are soil-borne fungal pathogens for Legumes
GWAS workshop
Verticillium sp. are soil-borne fungal pathogens for Legumes
GWAS workshop
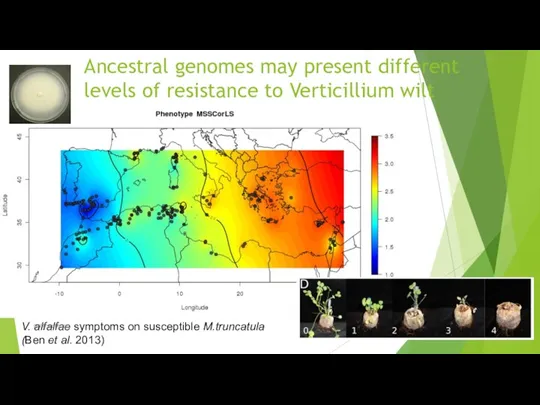
Слайд 80Ancestral genomes may present different levels of resistance to Verticillium wilt
V. alfalfae
Ancestral genomes may present different levels of resistance to Verticillium wilt
V. alfalfae
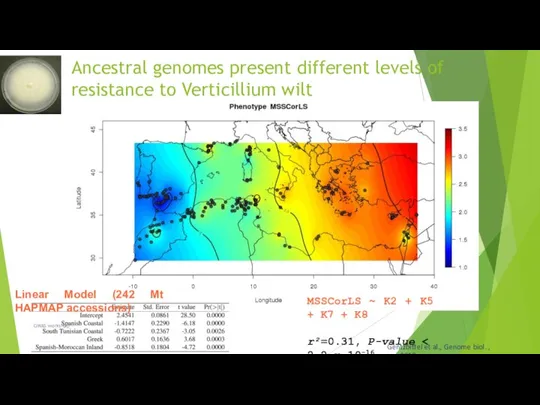
Слайд 81Ancestral genomes present different levels of resistance to Verticillium wilt
Linear Model (242
Ancestral genomes present different levels of resistance to Verticillium wilt
Linear Model (242
Слайд 82Can phenotype be predicted using WhoGem?
Use plant model (Medicago truncatula) to test
Can phenotype be predicted using WhoGem?
Use plant model (Medicago truncatula) to test

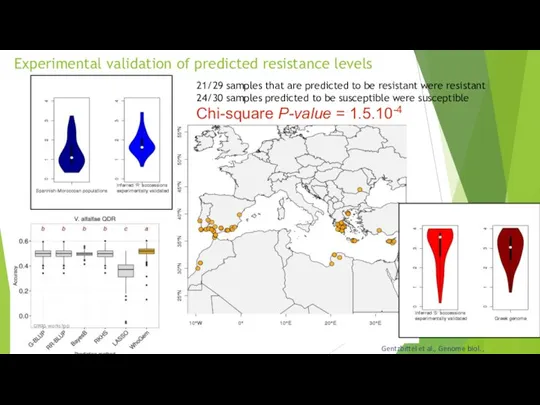
Слайд 83Experimental validation of predicted resistance levels
21/29 samples that are predicted to be
Experimental validation of predicted resistance levels
21/29 samples that are predicted to be
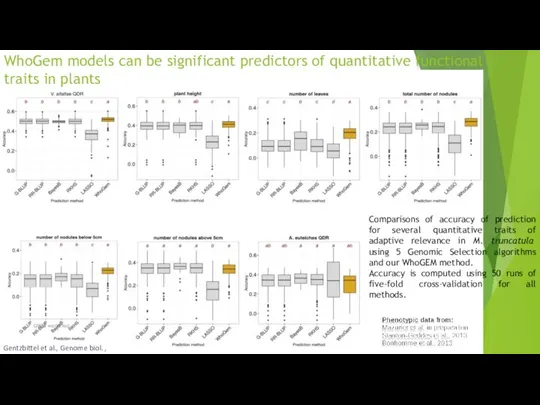
Слайд 84WhoGem models can be significant predictors of quantitative functional traits in plants
Comparisons
WhoGem models can be significant predictors of quantitative functional traits in plants
Comparisons
Слайд 85What is missing?
Distance between two points should not be just geometric distance.
What is missing?
Distance between two points should not be just geometric distance.

Слайд 86GWAS workshop
GWAS workshop
Слайд 87GWAS workshop
GWAS workshop

Слайд 88GWAS workshop
GWAS workshop
Слайд 89This methodology may serve as a basis for analyses in other plant
This methodology may serve as a basis for analyses in other plant

Слайд 90Practice
Admixture.csv
Pheno.csv
GWAS workshop
Practice
Admixture.csv
Pheno.csv
GWAS workshop

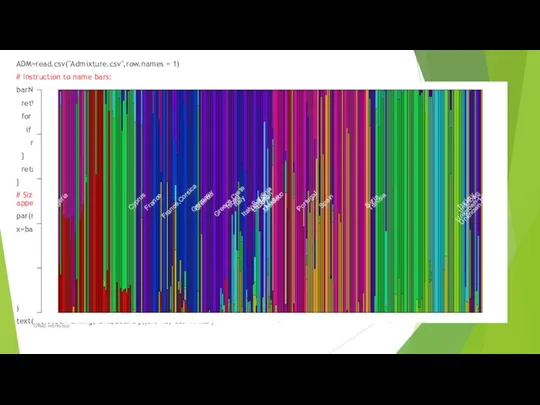
Слайд 91ADM=read.csv("Admixture.csv",row.names = 1)
# Instruction to name bars:
barNaming <- function(vec) {
retVec <-
ADM=read.csv("Admixture.csv",row.names = 1)
# Instruction to name bars:
barNaming <- function(vec) {
retVec <-
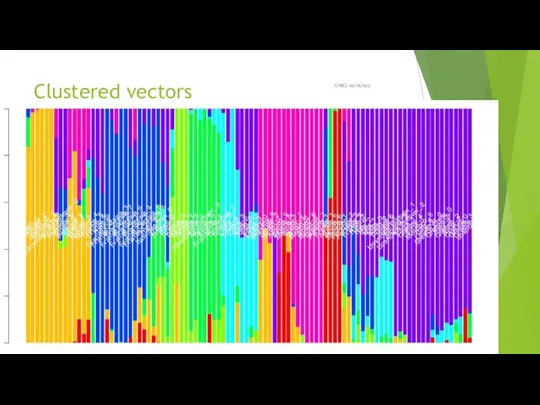
Слайд 92Clustered vectors
GWAS workshop
Clustered vectors
GWAS workshop
Слайд 93Merge datasets
ADM=read.csv('Admixture.csv',row.names = 1); TEST=subset(ADM, is.na(ADM$Lat))
REF=subset(ADM, !is.na(ADM$Lat)); Phen=read.csv('pheno.csv',row.names = 1); head(Phen)
Merge datasets
ADM=read.csv('Admixture.csv',row.names = 1); TEST=subset(ADM, is.na(ADM$Lat))
REF=subset(ADM, !is.na(ADM$Lat)); Phen=read.csv('pheno.csv',row.names = 1); head(Phen)

Слайд 94DATA=AMDPH[,c(4:6,8:15,17)];
cor(DATA)
GWAS workshop
DATA=AMDPH[,c(4:6,8:15,17)];
cor(DATA)
GWAS workshop
![DATA=AMDPH[,c(4:6,8:15,17)]; cor(DATA) GWAS workshop](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1062822/slide-93.jpg)
Слайд 95##GPS
M=25;
for(i in 1:dim(TEST)[1]){
Y=TEST[i,7:14];Y
DIST=c()
for(j in 1:dim(AMDPH)[1]){
Z=AMDPH[j,8:15];Z
d=sum((Y-Z)^2);d
##GPS
M=25;
for(i in 1:dim(TEST)[1]){
Y=TEST[i,7:14];Y
DIST=c()
for(j in 1:dim(AMDPH)[1]){
Z=AMDPH[j,8:15];Z
d=sum((Y-Z)^2);d
![##GPS M=25; for(i in 1:dim(TEST)[1]){ Y=TEST[i,7:14];Y DIST=c() for(j in 1:dim(AMDPH)[1]){ Z=AMDPH[j,8:15];Z d=sum((Y-Z)^2);d](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1062822/slide-94.jpg)
Слайд 96Results
GWAS workshop
Results
GWAS workshop
Слайд 97#GLM
library(lmtest); library(dplyr); library(car); library(rcompanion)
#set up formulas
formula=formula(MaxSymptomScore~K1+K2+K3+K4+K5+K6+K7+K8)
formula0=MaxSymptomScore ~ 1
#initialize models
model.null = glm(formula0, data=AMDPH);
#GLM
library(lmtest); library(dplyr); library(car); library(rcompanion)
#set up formulas
formula=formula(MaxSymptomScore~K1+K2+K3+K4+K5+K6+K7+K8)
formula0=MaxSymptomScore ~ 1
#initialize models
model.null = glm(formula0, data=AMDPH);

Слайд 98Laurent Gentzbittel, Ecolab, Toulouse, France/Skoltech, Moscow, Russia
Nevin Young's lab, Univ. of Minnesota,
Laurent Gentzbittel, Ecolab, Toulouse, France/Skoltech, Moscow, Russia
Nevin Young's lab, Univ. of Minnesota,

 Хордовые: строение, происхождение и родственные связи
Хордовые: строение, происхождение и родственные связи Растительные объекты,которые надо знать
Растительные объекты,которые надо знать Теории эволюции
Теории эволюции Зрительный анализатор. Урок 13
Зрительный анализатор. Урок 13 Братья наши меньшие
Братья наши меньшие Забота о потомстве
Забота о потомстве Как мыслит собака
Как мыслит собака Первоцветы села Казьминское
Первоцветы села Казьминское Внутреннее строение листа растения
Внутреннее строение листа растения Онтогенез. Эмбриология – наука о развитии организмов
Онтогенез. Эмбриология – наука о развитии организмов Тип членистоногие. Классы Ракообразные, Паукообразные, Насекомые
Тип членистоногие. Классы Ракообразные, Паукообразные, Насекомые Метаболизм углеводов. Энергетический обмен
Метаболизм углеводов. Энергетический обмен Строение скелета человека. Значение скелета для жизнедеятельности организма
Строение скелета человека. Значение скелета для жизнедеятельности организма Презентация на тему Органические вещества живых организмов
Презентация на тему Органические вещества живых организмов  Митоз бен мейоздың ұқсастықтары мен айырмашылықтары
Митоз бен мейоздың ұқсастықтары мен айырмашылықтары Патофизиология клетки
Патофизиология клетки Половое размножение цветковых растений
Половое размножение цветковых растений Круглые черви. (7)
Круглые черви. (7) Цитоплазма растительной клетки
Цитоплазма растительной клетки Реализация наследственной информации в клетке
Реализация наследственной информации в клетке Жизненный цикл бабочки
Жизненный цикл бабочки Эндокринные железы
Эндокринные железы חיות בעולם
חיות בעולם Анализаторы, строение и функции
Анализаторы, строение и функции Огуречные секреты
Огуречные секреты Виды стебля
Виды стебля Введение в предмет. Уровни организации живого организма
Введение в предмет. Уровни организации живого организма Генетика. Эпигенетические изменения
Генетика. Эпигенетические изменения