- Множественные выравнивания

Содержание
- 2. Что такое множественное выравнивание? Несколько гомологичных последовательностей, написанных друг под другом оптимальным способом: Гомологичные остатки один
- 3. Какое выравнивание интереснее?
- 4. Какие бывают выравнивания? локальные глобальные локальные глобальные множественные парные Выравнивания
- 5. Зачем нужно множественное выравнивание? Перенос аннотации Предсказание функции каждого остатка (например, выявление остатков, составляющих активный центр
- 6. Как выбрать последовательности для множественного выравнивания? Выравнивайте белки, а не ДНК, если есть выбор Последовательностей лучше
- 7. Изучая новую последовательность Выборка на основе BLAST Подробно охарактеризованные последовательности - аннотация Совсем неохарактеризованные (hypothetical proteins)
- 8. Подготовка выборки BLAST => сохранить все последовательности разом в FASTA формате или сразу на выравнивание Имена
- 9. Как можно строить глобальное множественное выравнивание? Построение множественного выравнивания N последовательностей t =LN !!! Можно пытаться
- 10. Алгоритм ClustalW – пример эвристического прогрессивного алгоритма Руководящее дерево Очевидные недостатки: Результат зависит от порядка выравниваний;
- 11. Современные методы построения множественного выравнивания (MSA, multiple sequence alignment): Алгоритм ClustalW (реализации ClustalX, emma из EMBOSS)
- 12. Использование ClustalW
- 13. Какие output-форматы бывают Post-script, pdf, html – только графика FASTA – последовательности отдельно, но с пробелами
- 14. Перевод форматов: READSEQ (http://www-bimas.cit.nih.gov/molbio/readseq/) Аналогично: SEQCHECK
- 15. ClustalW - output
- 16. JalView – редактирование выравниваний Другие программы для редактирования выравниваний (stand-alone): GeneDoc; CINEMA; Seaview; Belvu; Bioedit; DCSE
- 17. TCoffee Построение множественных выравниваний Оценка достоверности существующего выравнивания Использование 3-D структуры при построении выравнивания Сравнение и
- 18. TCoffee Выход – файлы clustalw_alnВыход – файлы clustalw_aln, fasta_alnВыход – файлы clustalw_aln, fasta_aln, phylipВыход – файлы
- 19. Как использовать TCoffee для других целей Множественное выравнивание на основе 3D-структуры (Expresso): надо заменить 1 или
- 20. Как “читать” множественное выравнивание? Хорошее выравнивание – высоко-консервативные блоки, перемежающиеся блоками с инсерциями/делециями ДНК – консервативные
- 21. Если консервативны только отдельные столбцы W, Y, F – консервативное гидрофобное ядро, стабилизирующая роль в ядре.
- 22. Локальное множественное выравнивание – постановка задачи Ряд последовательностей, в каждой из которых есть интересное слово (либо
- 23. dnaN ACATTATCCGTTAGGAGGATAAAAATG gyrA GTGATACTTCAGGGAGGTTTTTTAATG serS TCAATAAAAAAAGGAGTGTTTCGCATG bofA CAAGCGAAGGAGATGAGAAGATTCATG csfB GCTAACTGTACGGAGGTGGAGAAGATG xpaC ATAGACACAGGAGTCGATTATCTCATG metS ACATTCTGATTAGGAGGTTTCAAGATG gcaD AAAAGGGATATTGGAGGCCAATAAATG
- 24. Gibbs sampler Let’s A be a signal (set of sites), and I(A) be its information content.
- 25. Соответствующие программы
- 26. Представление результатов таких программ – Logos Программы построения – http://www-lmmb.ncifcrf.gov/~toms/sequencelogo.html; http://www.cbs.dtu.dk/~gorodkin/appl/plogo.html
- 27. Greedy algorithms (MEME) Find a signal among all k-words (assuming that we know the length signal).
- 28. Greedy algorithms. Cont’d Select the k-word with maximal information content Problem. We considered only k-words from
- 29. Limitation of greedy algorithms Started from k-words in our sequences and increase the information content at
- 31. Скачать презентацию
Слайд 2Что такое множественное выравнивание?
Несколько гомологичных последовательностей, написанных друг под другом оптимальным способом:
Гомологичные
Что такое множественное выравнивание?
Несколько гомологичных последовательностей, написанных друг под другом оптимальным способом:
Гомологичные

Слайд 3Какое выравнивание интереснее?
Какое выравнивание интереснее?

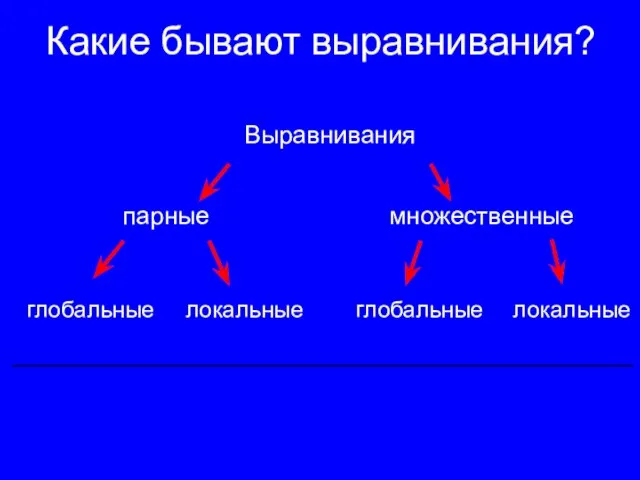
Слайд 4Какие бывают выравнивания?
локальные
глобальные
локальные
глобальные
множественные
парные
Выравнивания
Какие бывают выравнивания?
локальные
глобальные
локальные
глобальные
множественные
парные
Выравнивания
Слайд 5Зачем нужно множественное выравнивание?
Перенос аннотации
Предсказание функции каждого остатка (например, выявление остатков, составляющих
Зачем нужно множественное выравнивание?
Перенос аннотации
Предсказание функции каждого остатка (например, выявление остатков, составляющих

Слайд 6Как выбрать последовательности для множественного выравнивания?
Выравнивайте белки, а не ДНК, если есть
Как выбрать последовательности для множественного выравнивания?
Выравнивайте белки, а не ДНК, если есть

Слайд 7Изучая новую последовательность
Выборка на основе BLAST
Подробно охарактеризованные последовательности - аннотация
Совсем неохарактеризованные (hypothetical
Изучая новую последовательность
Выборка на основе BLAST
Подробно охарактеризованные последовательности - аннотация
Совсем неохарактеризованные (hypothetical

Слайд 8Подготовка выборки
BLAST => сохранить все последовательности разом в FASTA формате или сразу
Подготовка выборки
BLAST => сохранить все последовательности разом в FASTA формате или сразу

Слайд 9Как можно строить глобальное множественное выравнивание?
Построение множественного выравнивания N последовательностей
t =LN !!!
Можно
Как можно строить глобальное множественное выравнивание?
Построение множественного выравнивания N последовательностей
t =LN !!!
Можно

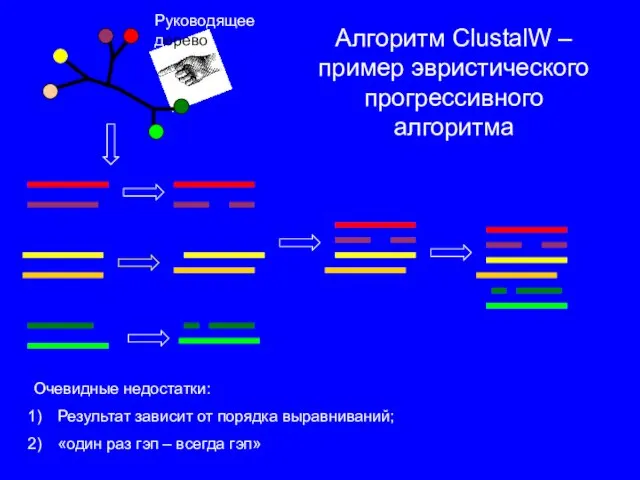
Слайд 10Алгоритм ClustalW – пример эвристического прогрессивного алгоритма
Руководящее дерево
Очевидные недостатки:
Результат зависит от
Алгоритм ClustalW – пример эвристического прогрессивного алгоритма
Руководящее дерево
Очевидные недостатки:
Результат зависит от
Слайд 11Современные методы построения множественного выравнивания
(MSA, multiple sequence alignment):
Алгоритм ClustalW (реализации ClustalX,
Современные методы построения множественного выравнивания
(MSA, multiple sequence alignment):
Алгоритм ClustalW (реализации ClustalX,

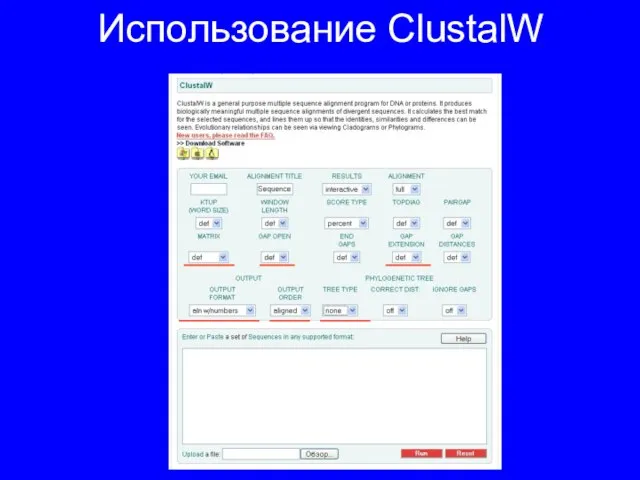
Слайд 12Использование ClustalW
Использование ClustalW
Слайд 13Какие output-форматы бывают
Post-script, pdf, html – только графика
FASTA – последовательности отдельно, но
Какие output-форматы бывают
Post-script, pdf, html – только графика
FASTA – последовательности отдельно, но

Слайд 14Перевод форматов: READSEQ
(http://www-bimas.cit.nih.gov/molbio/readseq/)
Аналогично: SEQCHECK
Перевод форматов: READSEQ
(http://www-bimas.cit.nih.gov/molbio/readseq/)
Аналогично: SEQCHECK
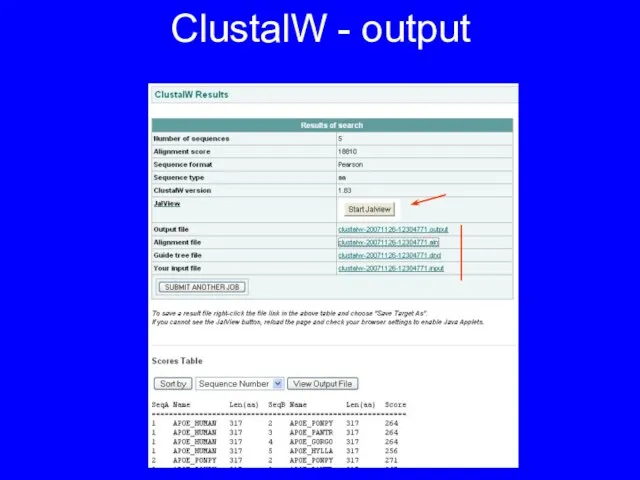
Слайд 15ClustalW - output
ClustalW - output
Слайд 16JalView – редактирование выравниваний
Другие программы для редактирования выравниваний (stand-alone):
GeneDoc; CINEMA; Seaview; Belvu;
JalView – редактирование выравниваний
Другие программы для редактирования выравниваний (stand-alone):
GeneDoc; CINEMA; Seaview; Belvu;

Слайд 17TCoffee
Построение множественных выравниваний
Оценка достоверности существующего выравнивания
Использование 3-D структуры при построении выравнивания
Сравнение и
TCoffee
Построение множественных выравниваний
Оценка достоверности существующего выравнивания
Использование 3-D структуры при построении выравнивания
Сравнение и

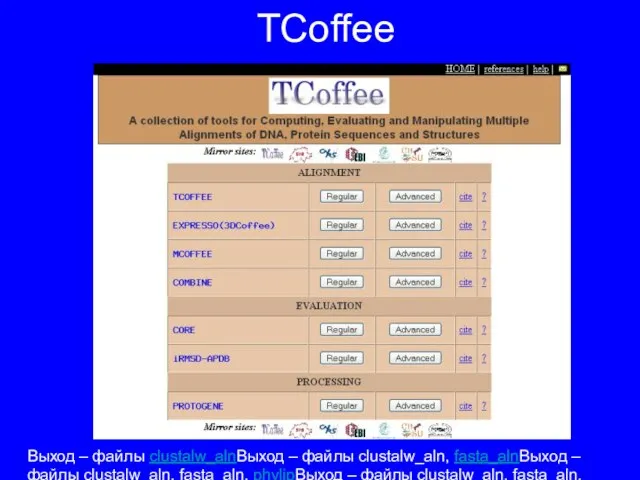
Слайд 18TCoffee
Выход – файлы clustalw_alnВыход – файлы clustalw_aln, fasta_alnВыход – файлы clustalw_aln, fasta_aln,
TCoffee
Выход – файлы clustalw_alnВыход – файлы clustalw_aln, fasta_alnВыход – файлы clustalw_aln, fasta_aln,
Слайд 19Как использовать TCoffee для других целей
Множественное выравнивание на основе 3D-структуры (Expresso): надо
Как использовать TCoffee для других целей
Множественное выравнивание на основе 3D-структуры (Expresso): надо

Слайд 20Как “читать” множественное выравнивание?
Хорошее выравнивание – высоко-консервативные блоки, перемежающиеся блоками с инсерциями/делециями
ДНК
Как “читать” множественное выравнивание?
Хорошее выравнивание – высоко-консервативные блоки, перемежающиеся блоками с инсерциями/делециями
ДНК

Слайд 21Если консервативны только отдельные столбцы
W, Y, F – консервативное гидрофобное ядро, стабилизирующая
Если консервативны только отдельные столбцы
W, Y, F – консервативное гидрофобное ядро, стабилизирующая

Слайд 22Локальное множественное выравнивание – постановка задачи
Ряд последовательностей, в каждой из которых есть
Локальное множественное выравнивание – постановка задачи
Ряд последовательностей, в каждой из которых есть

Слайд 23dnaN ACATTATCCGTTAGGAGGATAAAAATG
gyrA GTGATACTTCAGGGAGGTTTTTTAATG
serS TCAATAAAAAAAGGAGTGTTTCGCATG
bofA CAAGCGAAGGAGATGAGAAGATTCATG
csfB GCTAACTGTACGGAGGTGGAGAAGATG
xpaC ATAGACACAGGAGTCGATTATCTCATG
metS ACATTCTGATTAGGAGGTTTCAAGATG
gcaD AAAAGGGATATTGGAGGCCAATAAATG
spoVC TATGTGACTAAGGGAGGATTCGCCATG
ftsH GCTTACTGTGGGAGGAGGTAAGGAATG
pabB AAAGAAAATAGAGGAATGATACAAATG
rplJ
dnaN ACATTATCCGTTAGGAGGATAAAAATG
gyrA GTGATACTTCAGGGAGGTTTTTTAATG
serS TCAATAAAAAAAGGAGTGTTTCGCATG
bofA CAAGCGAAGGAGATGAGAAGATTCATG
csfB GCTAACTGTACGGAGGTGGAGAAGATG
xpaC ATAGACACAGGAGTCGATTATCTCATG
metS ACATTCTGATTAGGAGGTTTCAAGATG
gcaD AAAAGGGATATTGGAGGCCAATAAATG
spoVC TATGTGACTAAGGGAGGATTCGCCATG
ftsH GCTTACTGTGGGAGGAGGTAAGGAATG
pabB AAAGAAAATAGAGGAATGATACAAATG
rplJ

Слайд 24Gibbs sampler
Let’s A be a signal (set of sites), and I(A) be
Gibbs sampler
Let’s A be a signal (set of sites), and I(A) be

Слайд 25Соответствующие программы
Соответствующие программы

Слайд 26Представление результатов таких программ – Logos
Программы построения –
http://www-lmmb.ncifcrf.gov/~toms/sequencelogo.html;
http://www.cbs.dtu.dk/~gorodkin/appl/plogo.html
Представление результатов таких программ – Logos
Программы построения –
http://www-lmmb.ncifcrf.gov/~toms/sequencelogo.html;
http://www.cbs.dtu.dk/~gorodkin/appl/plogo.html

Слайд 27Greedy algorithms (MEME)
Find a signal among all k-words (assuming that we
Greedy algorithms (MEME)
Find a signal among all k-words (assuming that we

Слайд 28Greedy algorithms. Cont’d
Select the k-word with maximal information content
Problem. We considered
Greedy algorithms. Cont’d
Select the k-word with maximal information content
Problem. We considered

Слайд 29Limitation of greedy algorithms
Started from k-words in our sequences and increase
Limitation of greedy algorithms
Started from k-words in our sequences and increase

 Структура презентации
Структура презентации Исправление документов в текущем периоде
Исправление документов в текущем периоде Услуги и службы инфокоммуникаций
Услуги и службы инфокоммуникаций Создание и настройка меню приложения
Создание и настройка меню приложения Правовое просвещение пожилых людей. Лекция-семинар: Осторожно - мошенники!
Правовое просвещение пожилых людей. Лекция-семинар: Осторожно - мошенники! Роль материаловедения в графическом дизайне
Роль материаловедения в графическом дизайне Fox on a box
Fox on a box Защита информации
Защита информации Основы геоинформационного картографирования
Основы геоинформационного картографирования 8 - Nested loops
8 - Nested loops Лекция о научных публикациях
Лекция о научных публикациях Делегаты. Лямбда выражения. События. Лекция 6
Делегаты. Лямбда выражения. События. Лекция 6 Регистрация в учебной среде ИнфоДа Moodle
Регистрация в учебной среде ИнфоДа Moodle Моделирование случайных процессов в среде табличного процессора
Моделирование случайных процессов в среде табличного процессора Анимация перемещения в программе Adobe Photoshop CC. Ключи анимации
Анимация перемещения в программе Adobe Photoshop CC. Ключи анимации Внедрение ИИ – ботов в инфраструктуру городов
Внедрение ИИ – ботов в инфраструктуру городов Полезные ресурсы. Работа в Сanva
Полезные ресурсы. Работа в Сanva IRO
IRO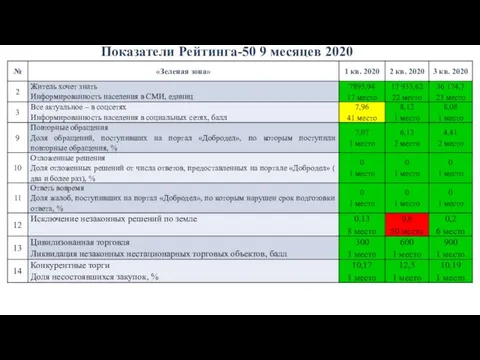 Показатели Рейтинга-50
Показатели Рейтинга-50 Новая технологическая платформа для метеообеспечения аэронавигации
Новая технологическая платформа для метеообеспечения аэронавигации ИДЗ. Алгоритм Дейкстры
ИДЗ. Алгоритм Дейкстры Работа журналиста с источниками информации
Работа журналиста с источниками информации Практическая работа Кодироваеие
Практическая работа Кодироваеие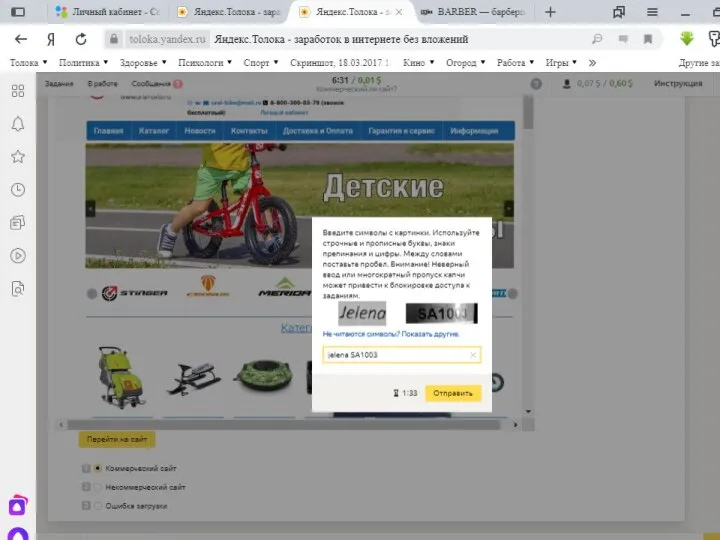 Скрин 1 на проверку
Скрин 1 на проверку Основные защитные механизмы, реализуемые в рамках различных мер и средств защиты
Основные защитные механизмы, реализуемые в рамках различных мер и средств защиты Разработка базы данных учета …
Разработка базы данных учета … Онлайн-обучение
Онлайн-обучение Основные угрозы и методы обеспечения информационной безопасности. Принципы защиты информации от несанкционированного доступа
Основные угрозы и методы обеспечения информационной безопасности. Принципы защиты информации от несанкционированного доступа