- Этическая экспертиза:нормативные документы,этапы прохождения

Содержание
- 2. ГЛАВНАЯ ЦЕЛЬ ЭТИЧЕСКОЙ ЭКСПЕРТИЗЫ Определить, с каким риском для субъекта исследования может быть связано их участие
- 3. НЕЗАВИСИМЫЙ ЭТИЧЕСКИЙ КОМИТЕТ Эта структура создается и существует именно для того, чтобы проводить этическую экспертизу Это
- 4. Обязательное требование GCP: Каждый исследовательский проект может осуществляться только после того, как независимым этическим комитетом будет
- 5. ЧТО НЕОБХОДИМО ЗНАТЬ? До начала исследования исследователь должен получить письменное одобрение этического комитета письменной формы информированного
- 6. ЭТАПЫ ПРОХОЖДЕНИЯ ЭТИЧЕСКОЙ ЭКСПЕРТИЗЫ
- 7. 1. Представление документов Документация по планируемому клиническому испытанию подается в Этический комитет от лица заявителя, несущего
- 8. 2. Сроки и адрес представления Документы подаются в Этический комитет СибГМУ каждый понедельник месяца с 14.00
- 9. Общие требования к предоставлению документов: Материалы исследования должны направляться в Этический комитет СибГМУ на имя председателя
- 10. Титульный лист 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
- 11. Титульный лист (на бумажную папку с завязками): Название протокола: Название исследования (должно соответствовать протоколу и аннотации)
- 12. 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ: 2.Подписанное заявителем и датированное заявление на рассмотрение Заявление
- 13. Заявка на проведение этической экспертизы Председателю Комитета по Этике СибГМУ д.м.н., профессору Е.Б.Букреевой Дата подачи документов
- 14. 3.Протокол планируемого исследования вместе с необходимыми приложениями и сопутствующими документами 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА
- 15. ПРОТОКОЛ ИССЛЕДОВАНИЯ Введение (краткое описание проблемы и схемы лечения) Цели исследования Длительность исследования Количество испытуемых Информированное
- 16. 4.Аннотация планируемого научного исследования 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
- 17. АННОТАЦИЯ НАУЧНОГО ИССЛЕДОВАНИЯ Требования стандартные для диссертационных работ
- 18. 5. Форма информационного листка субъекта исследования 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
- 19. ФОРМА ИНФОРМАЦИОННОГО ЛИСТКА
- 20. ЗАЧЕМ ИНФОРМАЦИОННЫЙ ЛИСТОК НУЖЕН ПАЦИЕНТУ Оно дает возможность участнику исследования ознакомиться с тем, … что его
- 21. РОЛЬ ИНФОРМАЦИОННОГО ЛИСТКА В КЛИНИЧЕСКОМ ИССЛЕДОВАНИИ Правильно заполненный информационный листок подтверждает правомерность участия пациента в клиническом
- 22. ПРОЦЕДУРА ПОЛУЧЕНИЯ ИНФОРМАЦИОННОГО ЛИСТКА Наличие времени для ознакомления Возможность ознакомиться с информационным листком субъекта или лиц,
- 23. ТРЕБОВАНИЯ К ФОРМЕ ИНФОРМАЦИОННОГО ЛИСТКА Текст на понятном пациенту языке Исключение использования не понятной пациенту терминологии
- 24. ТРЕБОВАНИЯ К ФОРМЕ ИНФОРМАЦИОННОГО ЛИСТКА При наличии специальных терминов необходима расшифровка или объяснение в тексте Шрифт
- 25. ОСНОВНЫЕ ЭЛЕМЕНТЫ ИНФОРМАЦИОННОГО ЛИСТКА Утверждение о статусе лечения Описание целей исследования Описание возможного риска Описание ожидаемой
- 26. Форма Информационного листка
- 27. 6. Информированное согласие 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
- 28. ИНФОРМИРОВАННОЕ СОГЛАСИЕ Это процесс, в ходе которого субъект исследования добровольно подтверждает свою волю участвовать в данном
- 29. ИНФОРМИРОВАННОЕ СОГЛАСИЕ Обеспечивает отношение к пациенту как к личности, которая вправе осуществлять свободный выбор и контролировать
- 30. ЦЕЛЬ ПОЛУЧЕНИЯ ИНФОРМИРОВАННОГО СОГЛАСИЯ Минимизировать возможность морального или материального ущерба вследствие недобросовестного лечения Способствовать повышению чувства
- 31. ПРОЦЕДУРА ПОЛУЧЕНИЯ ИНФОРМИРОВАННОГО СОГЛАСИЯ Это процесс ОБМЕНА ИНФОРМАЦИЕЙ, который имеет место между пациентом и исследователем –
- 32. КОГДА ИНФОРМИРОВАННОЕ СОГЛАСИЕ ДОЛЖНО БЫТЬ ПОДПИСАНО? Подписано до момента включения субъекта в исследование
- 33. КОГДА ИНФОРМИРОВАННОЕ СОГЛАСИЕ МОЖНО НЕ ПОДПИСЫВАТЬ? Процедуры, которые являются рутинными и предоставляются пациенту в обычном порядке
- 34. Типовые формы информированного согласия Настоящим я ___________________________________________________________ даю согласие на участие в испытании (название исследуемого препарата
- 35. ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ В некоторых случаях, если невозможно получить согласие пациента до начала исследования, возможна
- 36. ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ КОГДА ЭТО НУЖНО? Неотложные реанимационные состояния (инсульт, тяжелая травма и т.д.) В
- 37. ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ Если пациент включен в испытание с согласия его законного представителя, он должен
- 38. ПРИВЛЕЧЕНИЕ НЕЗАВИСИМЫХ СВИДЕТЕЛЕЙ КОГДА ЭТО НУЖНО? Пациент слепой Пациент неграмотный Перевод невозможен КТО ИМ МОЖЕТ БЫТЬ?
- 39. ПРИВЛЕЧЕНИЕ НЕЗАВИСИМЫХ СВИДЕТЕЛЕЙ Свидетель НЕ ДАЕТ согласия от лица испытуемого Свидетель подписывает и датирует Свидетель подтверждает,
- 40. «УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ Лица, на желание которых участвовать в клинических исследованиях может повлиять ожидание каких-либо выгод от
- 41. «УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ Студенты медицинских, фармакологических, сестринских учебных заведений Младший персонал больниц и лабораторий, военнослужащие, заключенные Несовершеннолетние
- 42. «УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ Испытания педиатрических препаратов: «… согласие ребенка должно быть получено в дополнение к разрешению его
- 43. РАСПРОСТРАНЕННЫЕ ОШИБКИ Неполучение согласия до начала участия испытуемого в исследовании Не поставленные должным образом подписи и
- 44. 7. СПРАВКА О ВЫПОЛНЕННОМ ОБЪЕМЕ НАУЧНО-ИССЛЕДОВАТЕЛЬСКОЙ РАБОТЫ 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
- 45. СПРАВКА О ВЫПОЛНЕННОМ ОБЪЕМЕ Информирует Вас о том, что по протоколу Научно-исследовательской работы: _____________________________________________ на соискание
- 46. 8. ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ КАРТА/ФОРМА (ИРК/ИРФ) 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
- 47. ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ КАРТА / ФОРМА (ИРК / ИРФ) Это бумажный или электронный документ, предназначенный для внесения
- 48. ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ КАРТА / ФОРМА (ИРК / ИРФ) Должны быть указаны следующие данные: номер, название исследования
- 49. Должны быть предусмотрены страницы для внесения информации по следующим позициям: история болезни данные физического обследования основной
- 50. Исследователь несет ответственность за все данные, записанные в Индивидуальные регистрационные карты, и должен заверять их своей
- 51. 9. ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА 3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
- 52. ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА Название исследования Исполнитель (асп., докт., соиск. ) Руководитель исследования (консультант): Место проведения исследования: Цель
- 53. ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА (лист №1) Название исследования Исполнитель (асп., докт., соиск., исследователь) Руководитель исследования (консультант): Место проведения
- 54. ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА (лист №2) Исследование служит: Непосредственно интересам испытуемых Чисто научным целям, не имеющим непосредственного значения
- 55. 4. Оформление представления в Этическом комитете При получении папки всех документов проводится предварительная этическая экспертиза материалов
- 56. 4. Оформление представления в Этическом комитете Исследователь обязан быть на заседании Этического комитета Исследователь обязан в
- 58. Скачать презентацию
Слайд 2ГЛАВНАЯ ЦЕЛЬ
ЭТИЧЕСКОЙ ЭКСПЕРТИЗЫ
Определить, с каким риском для субъекта исследования может быть
ГЛАВНАЯ ЦЕЛЬ
ЭТИЧЕСКОЙ ЭКСПЕРТИЗЫ
Определить, с каким риском для субъекта исследования может быть

Слайд 3НЕЗАВИСИМЫЙ
ЭТИЧЕСКИЙ КОМИТЕТ
Эта структура создается и существует именно для того, чтобы
НЕЗАВИСИМЫЙ
ЭТИЧЕСКИЙ КОМИТЕТ
Эта структура создается и существует именно для того, чтобы

Слайд 4Обязательное требование GCP:
Каждый исследовательский проект может осуществляться
только после того,
как
Обязательное требование GCP:
Каждый исследовательский проект может осуществляться
только после того,
как

Слайд 5ЧТО НЕОБХОДИМО ЗНАТЬ?
До начала исследования исследователь должен получить письменное одобрение этического комитета
ЧТО НЕОБХОДИМО ЗНАТЬ?
До начала исследования исследователь должен получить письменное одобрение этического комитета

Слайд 6ЭТАПЫ ПРОХОЖДЕНИЯ
ЭТИЧЕСКОЙ ЭКСПЕРТИЗЫ
ЭТАПЫ ПРОХОЖДЕНИЯ
ЭТИЧЕСКОЙ ЭКСПЕРТИЗЫ

Слайд 71. Представление документов
Документация по планируемому клиническому испытанию подается в Этический комитет от
1. Представление документов
Документация по планируемому клиническому испытанию подается в Этический комитет от

Слайд 82. Сроки и адрес представления
Документы подаются в Этический комитет СибГМУ каждый понедельник
2. Сроки и адрес представления
Документы подаются в Этический комитет СибГМУ каждый понедельник

Слайд 9Общие требования к предоставлению документов:
Материалы исследования должны направляться в Этический комитет СибГМУ
Общие требования к предоставлению документов:
Материалы исследования должны направляться в Этический комитет СибГМУ

Слайд 10Титульный лист
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
Титульный лист
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:

Слайд 11Титульный лист
(на бумажную папку с завязками):
Название протокола: Название исследования
(должно соответствовать
Титульный лист
(на бумажную папку с завязками):
Название протокола: Название исследования
(должно соответствовать

Слайд 123. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
2.Подписанное заявителем и датированное заявление
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
2.Подписанное заявителем и датированное заявление

Слайд 13Заявка на проведение этической экспертизы
Председателю Комитета по Этике СибГМУ д.м.н., профессору Е.Б.Букреевой
Дата
Заявка на проведение этической экспертизы
Председателю Комитета по Этике СибГМУ д.м.н., профессору Е.Б.Букреевой
Дата

Слайд 143.Протокол планируемого исследования вместе с необходимыми приложениями и сопутствующими документами
3. ДОКУМЕНТАЦИЯ
3.Протокол планируемого исследования вместе с необходимыми приложениями и сопутствующими документами
3. ДОКУМЕНТАЦИЯ

Слайд 15ПРОТОКОЛ ИССЛЕДОВАНИЯ
Введение (краткое описание проблемы и схемы лечения)
Цели исследования
Длительность исследования
Количество испытуемых
Информированное
ПРОТОКОЛ ИССЛЕДОВАНИЯ
Введение (краткое описание проблемы и схемы лечения)
Цели исследования
Длительность исследования
Количество испытуемых
Информированное

Слайд 164.Аннотация планируемого научного исследования
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
4.Аннотация планируемого научного исследования
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:

Слайд 17АННОТАЦИЯ
НАУЧНОГО ИССЛЕДОВАНИЯ
Требования стандартные для диссертационных работ
АННОТАЦИЯ
НАУЧНОГО ИССЛЕДОВАНИЯ
Требования стандартные для диссертационных работ

Слайд 185. Форма информационного листка субъекта исследования
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА
5. Форма информационного листка субъекта исследования
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА

Слайд 19ФОРМА ИНФОРМАЦИОННОГО ЛИСТКА
ФОРМА ИНФОРМАЦИОННОГО ЛИСТКА

Слайд 20ЗАЧЕМ ИНФОРМАЦИОННЫЙ ЛИСТОК НУЖЕН ПАЦИЕНТУ
Оно дает возможность участнику исследования ознакомиться с тем,
ЗАЧЕМ ИНФОРМАЦИОННЫЙ ЛИСТОК НУЖЕН ПАЦИЕНТУ
Оно дает возможность участнику исследования ознакомиться с тем,

Слайд 21РОЛЬ
ИНФОРМАЦИОННОГО ЛИСТКА
В КЛИНИЧЕСКОМ ИССЛЕДОВАНИИ
Правильно заполненный информационный листок подтверждает правомерность участия
РОЛЬ
ИНФОРМАЦИОННОГО ЛИСТКА
В КЛИНИЧЕСКОМ ИССЛЕДОВАНИИ
Правильно заполненный информационный листок подтверждает правомерность участия

Слайд 22ПРОЦЕДУРА ПОЛУЧЕНИЯ ИНФОРМАЦИОННОГО ЛИСТКА
Наличие времени для ознакомления
Возможность ознакомиться с информационным листком субъекта
ПРОЦЕДУРА ПОЛУЧЕНИЯ ИНФОРМАЦИОННОГО ЛИСТКА
Наличие времени для ознакомления
Возможность ознакомиться с информационным листком субъекта

Слайд 23ТРЕБОВАНИЯ К ФОРМЕ ИНФОРМАЦИОННОГО ЛИСТКА
Текст на понятном пациенту языке
Исключение использования не
ТРЕБОВАНИЯ К ФОРМЕ ИНФОРМАЦИОННОГО ЛИСТКА
Текст на понятном пациенту языке
Исключение использования не

Слайд 24ТРЕБОВАНИЯ К ФОРМЕ ИНФОРМАЦИОННОГО ЛИСТКА
При наличии специальных терминов необходима расшифровка или
ТРЕБОВАНИЯ К ФОРМЕ ИНФОРМАЦИОННОГО ЛИСТКА
При наличии специальных терминов необходима расшифровка или

Слайд 25ОСНОВНЫЕ ЭЛЕМЕНТЫ ИНФОРМАЦИОННОГО ЛИСТКА
Утверждение о статусе лечения
Описание целей исследования
Описание возможного риска
Описание
ОСНОВНЫЕ ЭЛЕМЕНТЫ ИНФОРМАЦИОННОГО ЛИСТКА
Утверждение о статусе лечения
Описание целей исследования
Описание возможного риска
Описание

Слайд 26Форма Информационного листка
Форма Информационного листка
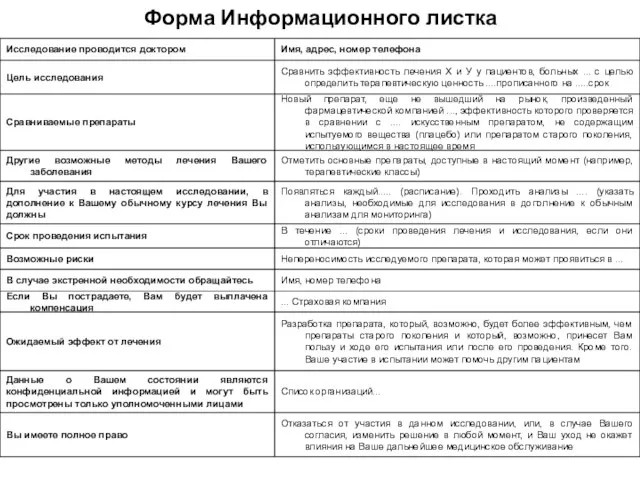
Слайд 276. Информированное согласие
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
6. Информированное согласие
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:

Слайд 28ИНФОРМИРОВАННОЕ СОГЛАСИЕ
Это процесс, в ходе которого субъект исследования добровольно подтверждает свою волю
ИНФОРМИРОВАННОЕ СОГЛАСИЕ
Это процесс, в ходе которого субъект исследования добровольно подтверждает свою волю

Слайд 29ИНФОРМИРОВАННОЕ СОГЛАСИЕ
Обеспечивает отношение к пациенту как к личности, которая вправе осуществлять свободный
ИНФОРМИРОВАННОЕ СОГЛАСИЕ
Обеспечивает отношение к пациенту как к личности, которая вправе осуществлять свободный

Слайд 30ЦЕЛЬ ПОЛУЧЕНИЯ ИНФОРМИРОВАННОГО СОГЛАСИЯ
Минимизировать возможность морального или материального ущерба вследствие недобросовестного лечения
Способствовать
ЦЕЛЬ ПОЛУЧЕНИЯ ИНФОРМИРОВАННОГО СОГЛАСИЯ
Минимизировать возможность морального или материального ущерба вследствие недобросовестного лечения
Способствовать

Слайд 31ПРОЦЕДУРА ПОЛУЧЕНИЯ ИНФОРМИРОВАННОГО СОГЛАСИЯ
Это процесс ОБМЕНА ИНФОРМАЦИЕЙ, который имеет место между пациентом
ПРОЦЕДУРА ПОЛУЧЕНИЯ ИНФОРМИРОВАННОГО СОГЛАСИЯ
Это процесс ОБМЕНА ИНФОРМАЦИЕЙ, который имеет место между пациентом

Слайд 32КОГДА
ИНФОРМИРОВАННОЕ СОГЛАСИЕ ДОЛЖНО БЫТЬ ПОДПИСАНО?
Подписано
до момента
включения субъекта
в исследование
КОГДА
ИНФОРМИРОВАННОЕ СОГЛАСИЕ ДОЛЖНО БЫТЬ ПОДПИСАНО?
Подписано
до момента
включения субъекта
в исследование

Слайд 33КОГДА
ИНФОРМИРОВАННОЕ СОГЛАСИЕ МОЖНО НЕ ПОДПИСЫВАТЬ?
Процедуры, которые являются рутинными и предоставляются пациенту
КОГДА
ИНФОРМИРОВАННОЕ СОГЛАСИЕ МОЖНО НЕ ПОДПИСЫВАТЬ?
Процедуры, которые являются рутинными и предоставляются пациенту

Слайд 34Типовые формы
информированного согласия
Настоящим я ___________________________________________________________ даю согласие на участие в
Типовые формы
информированного согласия
Настоящим я ___________________________________________________________ даю согласие на участие в

Слайд 35 ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ
В некоторых случаях, если невозможно получить согласие пациента до
ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ
В некоторых случаях, если невозможно получить согласие пациента до

Слайд 36 ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ
КОГДА ЭТО НУЖНО?
Неотложные реанимационные состояния (инсульт, тяжелая травма и
ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ
КОГДА ЭТО НУЖНО?
Неотложные реанимационные состояния (инсульт, тяжелая травма и

Слайд 37 ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ
Если пациент включен в испытание с согласия его законного
ПОЛУЧЕНИЕ СОГЛАСИЯ ЗАКОННОГО ПРЕДСТАВИТЕЛЯ
Если пациент включен в испытание с согласия его законного

Слайд 38 ПРИВЛЕЧЕНИЕ НЕЗАВИСИМЫХ СВИДЕТЕЛЕЙ
КОГДА ЭТО НУЖНО?
Пациент слепой
Пациент неграмотный
Перевод невозможен
КТО ИМ МОЖЕТ БЫТЬ?
Любой человек,
ПРИВЛЕЧЕНИЕ НЕЗАВИСИМЫХ СВИДЕТЕЛЕЙ
КОГДА ЭТО НУЖНО?
Пациент слепой
Пациент неграмотный
Перевод невозможен
КТО ИМ МОЖЕТ БЫТЬ?
Любой человек,

Слайд 39 ПРИВЛЕЧЕНИЕ
НЕЗАВИСИМЫХ СВИДЕТЕЛЕЙ
Свидетель НЕ ДАЕТ согласия от лица испытуемого
Свидетель подписывает и датирует
Свидетель
ПРИВЛЕЧЕНИЕ
НЕЗАВИСИМЫХ СВИДЕТЕЛЕЙ
Свидетель НЕ ДАЕТ согласия от лица испытуемого
Свидетель подписывает и датирует
Свидетель

Слайд 40«УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ
Лица, на желание которых участвовать в клинических исследованиях
может повлиять ожидание
«УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ
Лица, на желание которых участвовать в клинических исследованиях
может повлиять ожидание

Слайд 41«УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ
Студенты медицинских, фармакологических, сестринских учебных заведений
Младший персонал больниц и лабораторий, военнослужащие,
«УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ
Студенты медицинских, фармакологических, сестринских учебных заведений
Младший персонал больниц и лабораторий, военнослужащие,

Слайд 42«УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ
Испытания педиатрических препаратов:
«… согласие ребенка должно быть получено в дополнение
«УЯЗВИМЫЕ» ИСПЫТУЕМЫЕ
Испытания педиатрических препаратов:
«… согласие ребенка должно быть получено в дополнение

Слайд 43РАСПРОСТРАНЕННЫЕ ОШИБКИ
Неполучение согласия до начала участия испытуемого в исследовании
Не поставленные должным образом
РАСПРОСТРАНЕННЫЕ ОШИБКИ
Неполучение согласия до начала участия испытуемого в исследовании
Не поставленные должным образом

Слайд 447. СПРАВКА
О ВЫПОЛНЕННОМ ОБЪЕМЕ
НАУЧНО-ИССЛЕДОВАТЕЛЬСКОЙ РАБОТЫ
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА
7. СПРАВКА
О ВЫПОЛНЕННОМ ОБЪЕМЕ
НАУЧНО-ИССЛЕДОВАТЕЛЬСКОЙ РАБОТЫ
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА

Слайд 45СПРАВКА О ВЫПОЛНЕННОМ ОБЪЕМЕ
Информирует Вас о том, что по протоколу Научно-исследовательской работы:
_____________________________________________
на
СПРАВКА О ВЫПОЛНЕННОМ ОБЪЕМЕ
Информирует Вас о том, что по протоколу Научно-исследовательской работы:
_____________________________________________
на

Слайд 468. ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ
КАРТА/ФОРМА
(ИРК/ИРФ)
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
8. ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ
КАРТА/ФОРМА
(ИРК/ИРФ)
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:

Слайд 47ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ КАРТА / ФОРМА (ИРК / ИРФ)
Это бумажный или электронный документ,
ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ КАРТА / ФОРМА (ИРК / ИРФ)
Это бумажный или электронный документ,

Слайд 48ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ КАРТА / ФОРМА (ИРК / ИРФ)
Должны быть указаны следующие данные:
ИНДИВИДУАЛЬНАЯ РЕГИСТРАЦИОННАЯ КАРТА / ФОРМА (ИРК / ИРФ)
Должны быть указаны следующие данные:

Слайд 49Должны быть предусмотрены страницы для внесения информации по следующим позициям:
история болезни
Должны быть предусмотрены страницы для внесения информации по следующим позициям:
история болезни

Слайд 50Исследователь несет ответственность за все данные, записанные в Индивидуальные регистрационные карты, и
Исследователь несет ответственность за все данные, записанные в Индивидуальные регистрационные карты, и

Слайд 519. ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:
9. ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА
3. ДОКУМЕНТАЦИЯ В ЭТИЧЕСКИЙ КОМИТЕТ ДОЛЖНА ВКЛЮЧАТЬ СЛЕДУЮЩЕЕ:

Слайд 52ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА
Название исследования
Исполнитель (асп., докт., соиск. ) Руководитель исследования (консультант):
Место проведения исследования:
ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА
Название исследования
Исполнитель (асп., докт., соиск. ) Руководитель исследования (консультант):
Место проведения исследования:

Слайд 53ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА (лист №1)
Название исследования
Исполнитель (асп., докт., соиск., исследователь)
Руководитель исследования (консультант):
Место
ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА (лист №1)
Название исследования
Исполнитель (асп., докт., соиск., исследователь)
Руководитель исследования (консультант):
Место

Слайд 54ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА (лист №2)
Исследование служит:
Непосредственно интересам испытуемых
Чисто научным целям, не
ЗАКЛЮЧЕНИЕ РЕЦЕНЗЕНТА (лист №2)
Исследование служит:
Непосредственно интересам испытуемых
Чисто научным целям, не

Слайд 554. Оформление представления в Этическом комитете
При получении папки всех документов проводится предварительная
4. Оформление представления в Этическом комитете
При получении папки всех документов проводится предварительная

Слайд 564. Оформление представления в Этическом комитете
Исследователь обязан быть на заседании Этического комитета
Исследователь
4. Оформление представления в Этическом комитете
Исследователь обязан быть на заседании Этического комитета
Исследователь

 Психика человека
Психика человека ВОССТАНОВИТЕЛЬНОЕ ПРАВОСУДИЕ иЮВЕНАЛЬНЫЕ ТЕХНОЛОГИИ в РОССИИ
ВОССТАНОВИТЕЛЬНОЕ ПРАВОСУДИЕ иЮВЕНАЛЬНЫЕ ТЕХНОЛОГИИ в РОССИИ МЕЖВЕДОМСТВЕННОЕ ВЗАИМОДЕЙСТВИЕ ПРИ ОРГАНИЗАЦИИ СЕМЕЙНОГО УСТРОЙСТВА ДЕТЕЙ,ОСТАВШИХСЯБЕЗ ПОПЕЧЕНИЯ РОДИТЕЛЕЙ
МЕЖВЕДОМСТВЕННОЕ ВЗАИМОДЕЙСТВИЕ ПРИ ОРГАНИЗАЦИИ СЕМЕЙНОГО УСТРОЙСТВА ДЕТЕЙ,ОСТАВШИХСЯБЕЗ ПОПЕЧЕНИЯ РОДИТЕЛЕЙ Назначение и структура бизнес-плана
Назначение и структура бизнес-плана Тонкости продвижения интернет-магазиновв поисковых системах
Тонкости продвижения интернет-магазиновв поисковых системах Євроінтеграція України як чинник соціально-економічного розвитку держави. Роль освіти в розвитку партнерства України з іншими де
Євроінтеграція України як чинник соціально-економічного розвитку держави. Роль освіти в розвитку партнерства України з іншими де Особенности модернизации России – процесс взаимодействия инновационного и сырьевого векторов экономики Карпова Анна Владимиров
Особенности модернизации России – процесс взаимодействия инновационного и сырьевого векторов экономики Карпова Анна Владимиров Развитие психики человека
Развитие психики человека Сатиры А.Д.Кантемира
Сатиры А.Д.Кантемира Конкурсный проект смотровой площадки на вершине горы Машук
Конкурсный проект смотровой площадки на вершине горы Машук Вводный инструктаж. Формирование команды
Вводный инструктаж. Формирование команды Новая экономичная система импульсного пневмотранспорта порошкообразных сред
Новая экономичная система импульсного пневмотранспорта порошкообразных сред Подбор конфигурации и модернизация средств вычислительной техники
Подбор конфигурации и модернизация средств вычислительной техники Школьный Художественный музей
Школьный Художественный музей Прикладная геоэкология
Прикладная геоэкология Подвижная игрушка Слоненок
Подвижная игрушка Слоненок Я - мэр города Петрозаводска
Я - мэр города Петрозаводска Презентация на тему Иисус Христос – историческая личность или мифологический герой
Презентация на тему Иисус Христос – историческая личность или мифологический герой Культура Древнего Китая
Культура Древнего Китая Отраслевое административно-правовое регулирование в хозяйственно-экономических комплексах [часть 2]
Отраслевое административно-правовое регулирование в хозяйственно-экономических комплексах [часть 2] Свойства жидкостей, газов и твердых тел в пословицах
Свойства жидкостей, газов и твердых тел в пословицах Презентация на тему Богомол
Презентация на тему Богомол Временное трудоустройство. Подростки
Временное трудоустройство. Подростки Свобода. Уверенность. Выгода.
Свобода. Уверенность. Выгода. Комаров Сергей Приложения для социальных сетей. Использование приложений в качестве рекламных инструментов.
Комаров Сергей Приложения для социальных сетей. Использование приложений в качестве рекламных инструментов. Презентация "Религиозные праздники христиан" - скачать презентации по МХК
Презентация "Религиозные праздники христиан" - скачать презентации по МХК Оценка необходимости и обоснованности внедрения системы CRM в компании АБВ
Оценка необходимости и обоснованности внедрения системы CRM в компании АБВ Искусство быть здоровым.
Искусство быть здоровым.