Особенности оформления документов с целью замены регистрационного удостоверения на медицинское изделие
- Особенности оформления документов с целью замены регистрационного удостоверения на медицинское изделие

Содержание
- 2. Федеральная служба по надзору в сфере здравоохранения О порядке подготовки и оформления документов для целей внесения
- 3. Федеральная служба по надзору в сфере здравоохранения Особенности оформления документов с целью замены регистрационного удостоверения на
- 4. Замена регистрационного удостоверения на медицинское изделие Постановление Правительства Российской Федерации от 27.12.2012 № 1416 «Об утверждении
- 5. Регистрационные удостоверения на медицинское изделие, не подлежащие замене Регистрационные удостоверения, выданные после 1 января 2013 года
- 6. Документы, необходимые для осуществления процедуры замены регистрационного удостоверения Заявление на замену регистрационного удостоверения по форме, утвержденной
- 7. Наиболее часто встречающиеся основания для отказа в замене регистрационных удостоверений на медицинское изделие Некорректное оформление доверенности
- 8. Наиболее часто встречающиеся основания для отказа в замене регистрационных удостоверений на медицинское изделие Несоответствие сведений, указанных
- 9. Иные вопросы, возникающие при замене регистрационных удостоверений на медицинское изделие Процедура замены регистрационного удостоверения осуществляется Росздравнадзором
- 11. Федеральная служба по надзору в сфере здравоохранения Порядок подачи заявления и документов на внесение изменений в
- 12. Нормативно-правовые акты, применяемые при подготовке документов для процедуры внесения изменений в регистрационное удостоверение 1. Постановление Правительства
- 13. Государственная пошлина за процедуру внесения изменений в регистрационное удостоверение на медицинское изделие Согласно ст. 333.32.2 Налогового
- 14. Кто такой заявитель? Согласно пункту 8 Правил государственной регистрации, заявителем является: 1. Разработчик 2. Производитель медицинского
- 15. Оформление заявления о внесении изменений в регистрационное удостоверение на медицинское изделие Требования к заявлению определены п.9
- 16. п. 6 Административного регламента Федеральной службы по надзору в сфере здравоохранения по предоставлению государственной услуги по
- 17. Внесение изменений в регистрационное удостоверение согласно подпункту «а» п.37 Правил а) изменение сведений о заявителе (производителе):
- 18. Реорганизация юридического лица и (или) смена адреса места нахождения юридического лица Документы, подтверждающие такие изменения, например:
- 19. Изменение наименования заявителя (производителя) Письмо от производителя, в котором содержатся пояснения о характере внесенных изменений, а
- 20. Изменение юридического лица (другой производитель) документы от производителя, подтверждающие распределение ответственности за качество ранее произведенной продукции
- 21. Изменение адреса места производства медицинского изделия согласно подпункту «б» п.37 Правил государственной регистрации медицинских изделий документы,
- 22. Изменение наименования медицинского изделия согласно подпункту «в» п.37 Правил государственной регистрации медицинских изделий заявление с указанием
- 23. Изменение сведений о юридическом лице, на имя которого может быть выдано регистрационное удостоверение, согласно подпункту «г»
- 24. Указание вида медицинского изделия согласно подпункту «д» п.37 Правил заявление с указанием вида медицинского изделия сведения
- 25. Пункт 38 Правил государственной регистрации медицинских изделий Для внесения изменений в регистрационное удостоверение заявитель не позднее
- 26. Обращаем Внимание! При принятии решения о внесении изменений в регистрационное удостоверение регистрирующий орган оформляет и выдает
- 28. Федеральная служба по надзору в сфере здравоохранения Требования к документам, представляемым заявителем с целью получения дубликата
- 29. Выдача дубликата регистрационного удостоверения на медицинское изделие Правила государственной регистрации медицинских изделий, утвержденные постановлением Правительства Российской
- 30. Документы и сведения, предоставляемые для выдачи дубликата регистрационного удостоверения на медицинское изделие Заявление о выдаче дубликата
- 31. Доверенность от производителя медицинского изделия на выдачу дубликата регистрационного удостоверения с заверенным в установленном порядке переводом
- 32. Часто встречающиеся основания для отказа в выдаче дубликата регистрационного удостоверения на медицинское изделие Некорректное оформление доверенности
- 34. Федеральная служба по надзору в сфере здравоохранения Основные нарушения и недостатки при рассмотрении комплектов регистрационной документации
- 35. Нормативно-правовые акты, регламентирующие процедуры регистрации медицинских изделий и внесения изменений в регистрационные удостоверения на медицинские изделия
- 36. Государственная пошлина, предусмотренная налоговым законодательством Российской Федерации за процедуру внесения изменений в регистрационные документы Государственная пошлина
- 37. Внесение изменений в регистрационную документацию (п. 55 Правил) В случае необходимости (по желанию заявителя) внесения изменений
- 38. Внесение изменений в регистрационную документацию (п. 55 Правил) В случае необходимости внесения изменений в документы, указанные
- 39. Внесение изменений в регистрационную документацию (п. 55 Правил) Вне зависимости от причины внесения изменений всегда представляется
- 40. Внесение изменений в регистрационную документацию (п. 55 Правил) Основаниями для вынесения экспертным учреждением заключения о невозможности
- 41. В случае если внесение изменений в техническую и (или) эксплуатационную документацию касаются: Росздравнадзор самостоятельно (без проведения
- 42. Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил 1. Окончание срока действия технических
- 43. Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил 3. Изменение маркировки или упаковки
- 44. Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил 5. Изменение показаний (противопоказаний) к
- 46. Федеральная служба по надзору в сфере здравоохранения Требования к технической и эксплуатационной документации на медицинское изделие,
- 47. Необходимость предоставления технической и эксплуатационной документации Необходимость предоставления технической и эксплуатационной документации при внесении изменений в
- 48. Необходимость предоставления технической и эксплуатационной документации в рамках п. 37 Правил государственной регистрации медицинских изделий Пункт
- 49. Необходимость предоставления технической и эксплуатационной документации в рамках п. 55 Правил государственной регистрации медицинских изделий Пункт
- 50. Техническая документация производителя (изготовителя) Требования к технической документации установлены в пункте 4 Правил государственной регистрации медицинских
- 51. Эксплуатационная документация производителя (изготовителя) Требования к эксплуатационной документации установлены в пункте 4 Правил государственной регистрации медицинских
- 52. Возможные варианты изменений, требующие актуализации технической и эксплуатационной документации Изменение наименования изделия Сведения о неизменности функционального
- 53. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя) Основные несоответствия Изменилось функциональное назначение Расширились модельные и
- 54. Пример 1. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя) Аппарат лазерный терапевтический «Модель1» Разработана и
- 55. Пример 2. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя) Изменены функциональные характеристики и параметры (улучшены)
- 56. Пример 3. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя) МИ Изменение срока хранения Изменение срока
- 57. Основные рекомендации для производителя при внесении изменений по п.55 Правил государственной регистрации медицинских изделий документировать все
- 59. Федеральная служба по надзору в сфере здравоохранения Особенности оценки результатов технических испытаний при проведении экспертизы по
- 60. Пункт 55 - В случае необходимости (по желанию заявителя) внесения изменений в документы, предусмотренные подпунктом "а"
- 61. Пункт 4 Правил государственной регистрации медицинских изделий, утвержденных постановлением Правительства Российской Федерации от 27.12.2012 № 1416:
- 62. Оформление результатов технических испытаний для подтверждения внесенных изменений производится в соответствии с Приказ Минздрава России от
- 63. В п. 5 приказа Минздрава России от 09.01.2014 № 2н установлено: «Технические испытания медицинских изделий проводятся
- 64. Порядок проведения технических испытаний медицинских изделий при внесении изменений В рамках оценки и анализа данных проводятся:
- 65. Перечень документов, которые предоставляет Заявитель для проведения технических испытаний (п. 9 приказа Минздрава России от 09.01.2014
- 66. Акт результатов технических испытаний измененного медицинского изделия и Приложения к нему: фотографическое изображение общего вида медицинского
- 67. Перечень документов, подтверждающих результаты технических испытаний медицинских изделий при внесении изменений Акт Программа технических испытаний Фотографии
- 68. Перечень документов, подтверждающих результаты технических испытаний медицинских изделий при внесении изменений Акт оценки результатов технических испытаний
- 69. Структура Программы технических испытаний медицинских изделий при внесении изменений - вводная часть; - рассмотрение технической и
- 70. Структура и содержание протоколов испытаний (рекомендуемая) ГОСТ ИСО/МЭК 17025-2009 Общие требования к компетентности испытательных и калибровочных
- 71. Структура и содержание протоколов испытаний (рекомендуемая) - результаты проведенных испытаний медицинского изделия и его принадлежностей по
- 72. Распространенные замечания по результатам оценки протоколов технических испытаний внесенных изменений Испытания проведены на соответствие недействующим национальным
- 73. Рекомендации для проведения технических испытаний при внесении изменений в документацию Проверить наличие действующей аккредитации у лаборатории
- 75. Федеральная служба по надзору в сфере здравоохранения Особенности оценки результатов токсикологических исследований при проведении экспертизы по
- 76. Пункт 4 Правил государственной регистрации медицинских изделий, утвержденных постановлением Правительства Российской Федерации от 27.12.2012 № 1416:
- 77. Оформление результатов токсикологических исследований для подтверждения внесенных изменений производится в соответствии с Приказом Минздрава России от
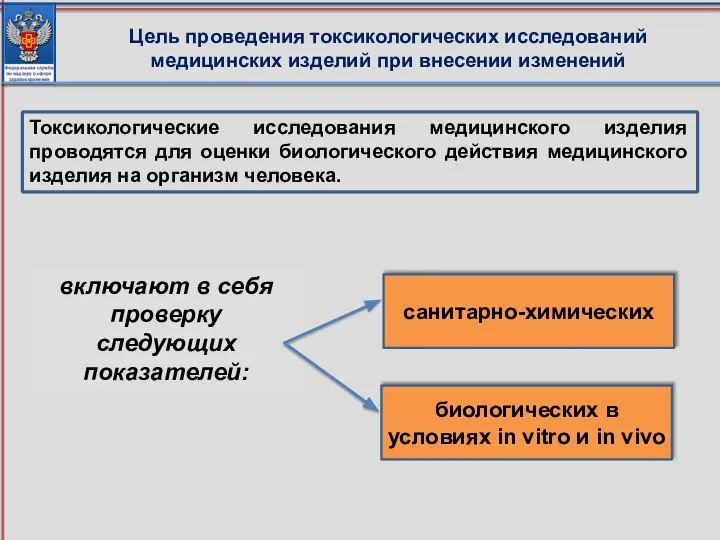
- 78. биологических в условиях in vitro и in vivo санитарно-химических Цель проведения токсикологических исследований медицинских изделий при
- 79. а) заявление о проведении токсикологических исследований; б) образцы (образец) медицинского изделия или принадлежности, к медицинскому изделию,
- 80. идентификация медицинского изделия (материала); классификация медицинского изделия; определение длительности контакта медицинского изделия с организмом человека; анализ
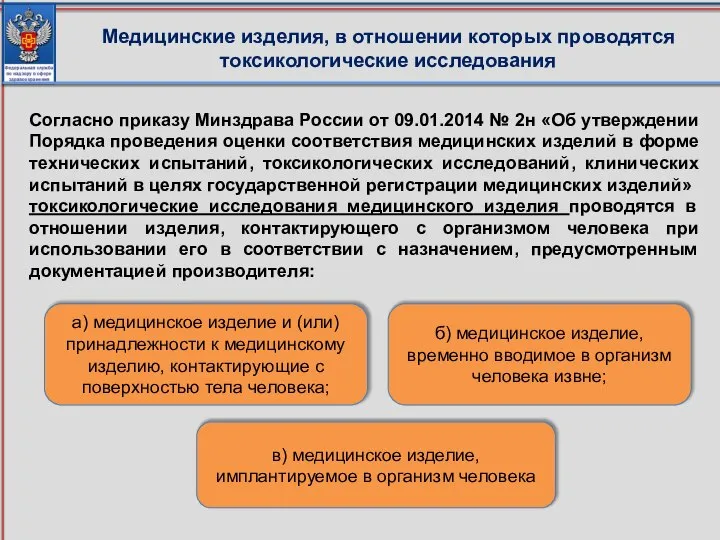
- 81. Медицинские изделия, в отношении которых проводятся токсикологические исследования Согласно приказу Минздрава России от 09.01.2014 № 2н
- 82. Медицинские изделия по виду контакта с организмом человека подразделяют на несколько групп. Изделия, контактирующие непосредственно или
- 83. Программа токсикологических исследований составляется испытательной организацией совместно с заявителем и утверждается руководителем испытательной организации, проводящей токсикологические
- 84. Соблюдение положений стандартов серии ISO 10993 "Оценка биологического действия медицинских изделий" позволит обеспечить системный подход к
- 85. 1 этап: Санитарно-химические исследования Санитарно-химические исследования позволяют отбраковывать непригодную к применению в клинической практике продукцию. Результаты
- 86. 2 этап: Токсикологические исследования испытания на стерильность и апирогенность исследуемого материала тесты in vitro на культурах
- 87. 2 этап: Токсикологические исследования - полноту и объективность установленных технической и эксплуатационной документацией производителя характеристик, подлежащих
- 88. ГОСТ 31214-2003, стандарты серии ГОСТ ISO 10993, ГОСТ Р 52770-2007, Единые санитарно-эпидемиологические и гигиенические требования к
- 89. Оформляется заключение по результатам токсикологических исследований медицинского изделия, форма которого приведена в приложении № 3 к
- 90. Исследования проведены не в полном объеме Сведения в результирующих документах не совпадают с документами производителя Исследования
- 91. Рекомендации для проведения токсикологических исследований при внесении изменений в документацию
- 93. Федеральная служба по надзору в сфере здравоохранения Особенности оценки результатов клинических испытаний при проведении экспертизы по
- 94. Нормативно-правовое регулирование при внесении изменений в регистрационное досье Порядок проведения оценки соответствия медицинских изделий в форме
- 95. Приложения и дополнения к Акту клинических испытаний Следует обратить особое внимание, что в качестве приложений Акт
- 96. Базы данных для поиска EMBASE – Escerpta Medica published by Elsevier CENTRAL – The Cochrane Central
- 97. Публикации о клинической эффективности медицинского изделия
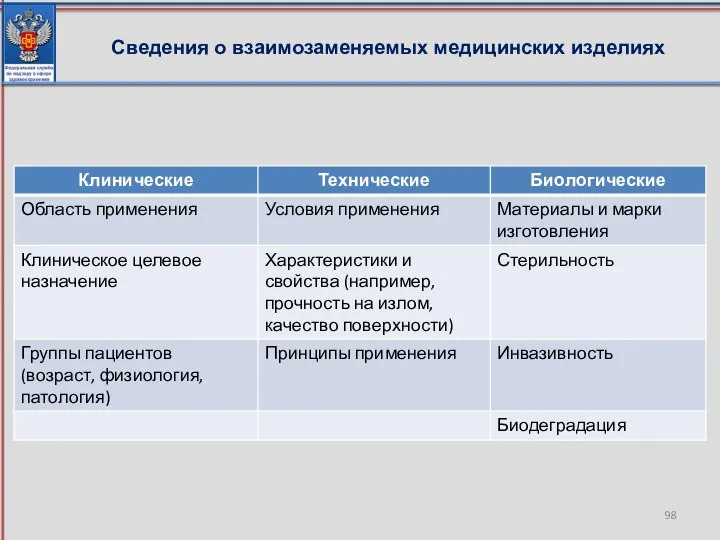
- 98. Сведения о взаимозаменяемых медицинских изделиях
- 99. Клинические испытания медицинских изделий для диагностики in vitro Клинические испытания медицинских изделий для диагностики in vitro
- 100. Результаты клинических испытаний считаются отрицательными: п. 45 Приказа Минздрава России от 09.01.2014 № 2н медицинское изделие
- 101. Основания для проведения клинических испытаний по п. 55 Правил государственной регистрации МИ Расширение показаний к применению
- 102. Пример 1. Расширение показаний к применению Изделие может использовать не только у взрослых, но и у
- 103. Пример 2. Уточнение/дополнение показаний к применению При неизменности функционального назначения и (или) принципа действия МИ Показания
- 104. Пример 3. Уточнение/дополнение противопоказаний к применению Противопоказания при регистрации МИ - Введение в область вокруг глаз
- 105. Пример 4. Уточнение/дополнение побочных эффектов при применении Новые побочные эффекты: - уплотнение или узелки в месте
- 106. Пример 5. Расширение области применения Области применения при регистрации: Шейный отдел позвоночника Запястье, кисть, пальцы Колени
- 107. Пример 6. Модернизация режима работы При неизменности функционального назначения и (или) принципа действия МИ Аппарат физиотерапевтический
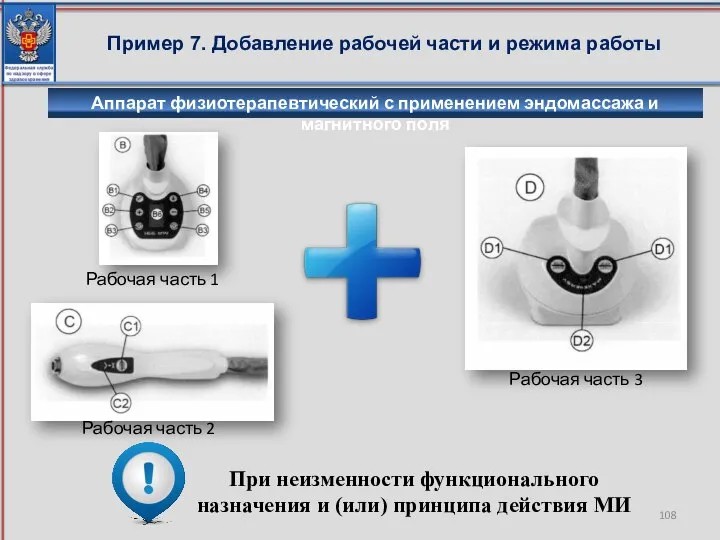
- 108. Пример 7. Добавление рабочей части и режима работы При неизменности функционального назначения и (или) принципа действия
- 109. Пример 8. Расширение условий применения При неизменности функционального назначения и (или) принципа действия МИ Тонометр Для
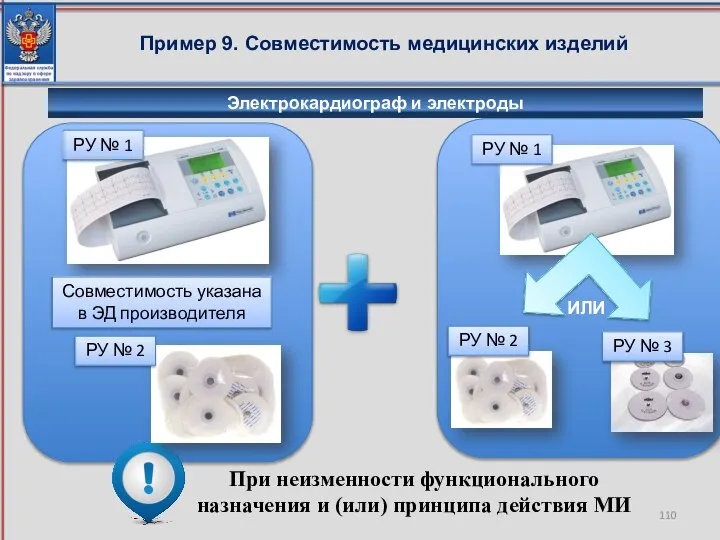
- 110. Пример 9. Совместимость медицинских изделий При неизменности функционального назначения и (или) принципа действия МИ РУ №
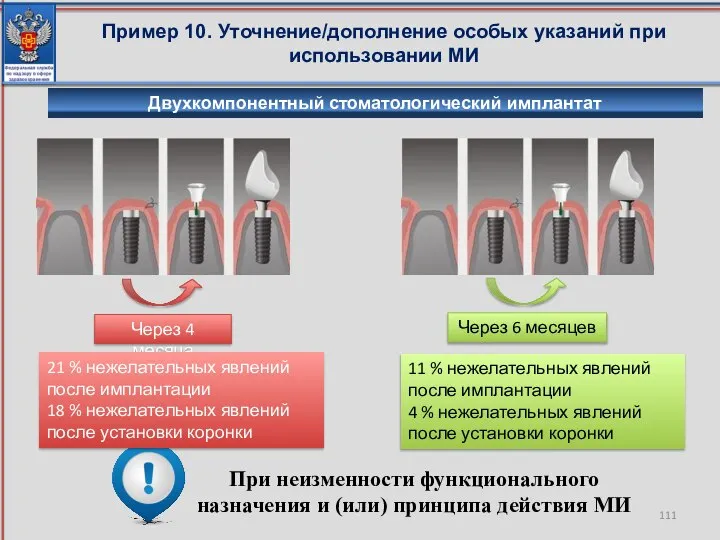
- 111. Пример 10. Уточнение/дополнение особых указаний при использовании МИ При неизменности функционального назначения и (или) принципа действия
- 112. Федеральная служба по надзору в сфере здравоохранения Анализ запросов о предоставлении дополнительных материалов и сведений и
- 113. п. 26: При внесении изменений в документы, указанные в подпунктах "в" и "г" пункта 10 Правил,
- 114. В случае недостаточности для вынесения экспертом заключения материалов и сведений, содержащихся в представленных заявителем заявлении о
- 115. Основаниями для вынесения экспертным учреждением заключения о невозможности внесения изменений в документы, предусмотренные подпунктами "в" и
- 116. Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий
- 117. Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий
- 118. Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий
- 119. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 120. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 121. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 122. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 123. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 124. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 125. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 126. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 127. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 128. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 129. Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной регистрации медицинских изделий
- 130. Федеральная служба по надзору в сфере здравоохранения Спасибо за внимание! [email protected]
- 132. Скачать презентацию
Слайд 2Федеральная служба по надзору в сфере здравоохранения
О порядке подготовки и оформления документов
Федеральная служба по надзору в сфере здравоохранения
О порядке подготовки и оформления документов

Слайд 3Федеральная служба по надзору в сфере здравоохранения
Особенности оформления документов с целью замены
Федеральная служба по надзору в сфере здравоохранения
Особенности оформления документов с целью замены

Слайд 4Замена регистрационного удостоверения
на медицинское изделие
Постановление Правительства Российской Федерации от 27.12.2012 № 1416
Замена регистрационного удостоверения
на медицинское изделие
Постановление Правительства Российской Федерации от 27.12.2012 № 1416

Слайд 5Регистрационные удостоверения на медицинское изделие,
не подлежащие замене
Регистрационные удостоверения, выданные после 1 января
Регистрационные удостоверения на медицинское изделие,
не подлежащие замене
Регистрационные удостоверения, выданные после 1 января

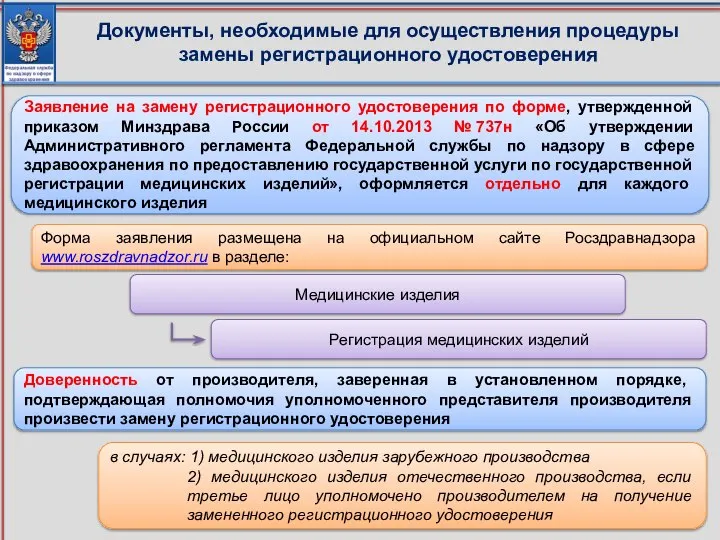
Слайд 6Документы, необходимые для осуществления процедуры замены регистрационного удостоверения
Заявление на замену регистрационного удостоверения
Документы, необходимые для осуществления процедуры замены регистрационного удостоверения
Заявление на замену регистрационного удостоверения
Слайд 7Наиболее часто встречающиеся основания для отказа в замене регистрационных удостоверений на медицинское
Наиболее часто встречающиеся основания для отказа в замене регистрационных удостоверений на медицинское

Слайд 8Наиболее часто встречающиеся основания для отказа в замене регистрационных удостоверений на медицинское
Наиболее часто встречающиеся основания для отказа в замене регистрационных удостоверений на медицинское

Слайд 9Иные вопросы, возникающие при замене регистрационных удостоверений на медицинское изделие
Процедура замены регистрационного
Иные вопросы, возникающие при замене регистрационных удостоверений на медицинское изделие
Процедура замены регистрационного


Слайд 11Федеральная служба по надзору в сфере здравоохранения
Порядок подачи заявления и документов на
Федеральная служба по надзору в сфере здравоохранения
Порядок подачи заявления и документов на

Слайд 12Нормативно-правовые акты, применяемые при подготовке документов для процедуры внесения изменений в регистрационное
Нормативно-правовые акты, применяемые при подготовке документов для процедуры внесения изменений в регистрационное

Слайд 13Государственная пошлина за процедуру внесения изменений в регистрационное удостоверение на медицинское изделие
Согласно
Государственная пошлина за процедуру внесения изменений в регистрационное удостоверение на медицинское изделие
Согласно

Слайд 14Кто такой заявитель?
Согласно пункту 8 Правил государственной регистрации, заявителем является:
1. Разработчик
2. Производитель
Кто такой заявитель?
Согласно пункту 8 Правил государственной регистрации, заявителем является:
1. Разработчик
2. Производитель

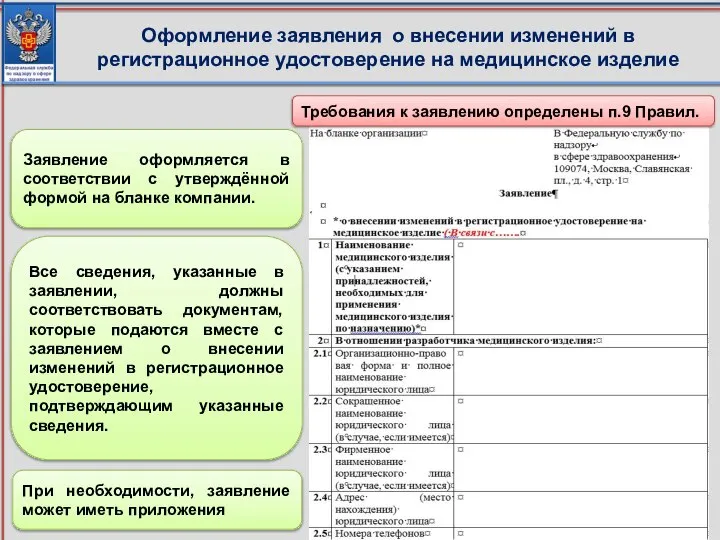
Слайд 15Оформление заявления о внесении изменений в регистрационное удостоверение на медицинское изделие
Требования к
Оформление заявления о внесении изменений в регистрационное удостоверение на медицинское изделие
Требования к
Слайд 16п. 6 Административного регламента Федеральной службы по надзору в сфере здравоохранения по
п. 6 Административного регламента Федеральной службы по надзору в сфере здравоохранения по

Слайд 17 Внесение изменений в регистрационное удостоверение согласно подпункту «а» п.37 Правил
а)
Внесение изменений в регистрационное удостоверение согласно подпункту «а» п.37 Правил
а)

Слайд 18Реорганизация юридического лица и (или) смена адреса места нахождения юридического лица
Документы, подтверждающие
Реорганизация юридического лица и (или) смена адреса места нахождения юридического лица
Документы, подтверждающие

Слайд 19Изменение наименования заявителя (производителя)
Письмо от производителя, в котором содержатся пояснения о характере
Изменение наименования заявителя (производителя)
Письмо от производителя, в котором содержатся пояснения о характере

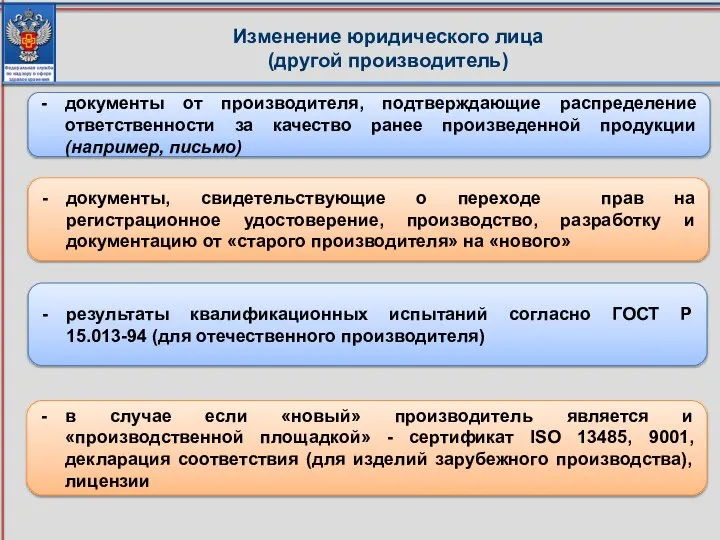
Слайд 20Изменение юридического лица
(другой производитель)
документы от производителя, подтверждающие распределение ответственности за качество
Изменение юридического лица
(другой производитель)
документы от производителя, подтверждающие распределение ответственности за качество
Слайд 21Изменение адреса места производства медицинского изделия согласно подпункту «б» п.37 Правил государственной
Изменение адреса места производства медицинского изделия согласно подпункту «б» п.37 Правил государственной

Слайд 22 Изменение наименования медицинского изделия
согласно подпункту «в» п.37 Правил государственной регистрации медицинских
Изменение наименования медицинского изделия согласно подпункту «в» п.37 Правил государственной регистрации медицинских

Слайд 23 Изменение сведений о юридическом лице, на имя которого может быть выдано
Изменение сведений о юридическом лице, на имя которого может быть выдано

Слайд 24 Указание вида медицинского изделия
согласно подпункту «д» п.37 Правил
заявление с указанием вида
Указание вида медицинского изделия
согласно подпункту «д» п.37 Правил
заявление с указанием вида

Слайд 25Пункт 38 Правил государственной регистрации медицинских изделий
Для внесения изменений в регистрационное удостоверение
Пункт 38 Правил государственной регистрации медицинских изделий
Для внесения изменений в регистрационное удостоверение

Слайд 26Обращаем Внимание!
При принятии решения о внесении изменений в регистрационное удостоверение регистрирующий орган
Обращаем Внимание!
При принятии решения о внесении изменений в регистрационное удостоверение регистрирующий орган


Слайд 28Федеральная служба по надзору в сфере здравоохранения
Требования к документам, представляемым заявителем с
Федеральная служба по надзору в сфере здравоохранения
Требования к документам, представляемым заявителем с

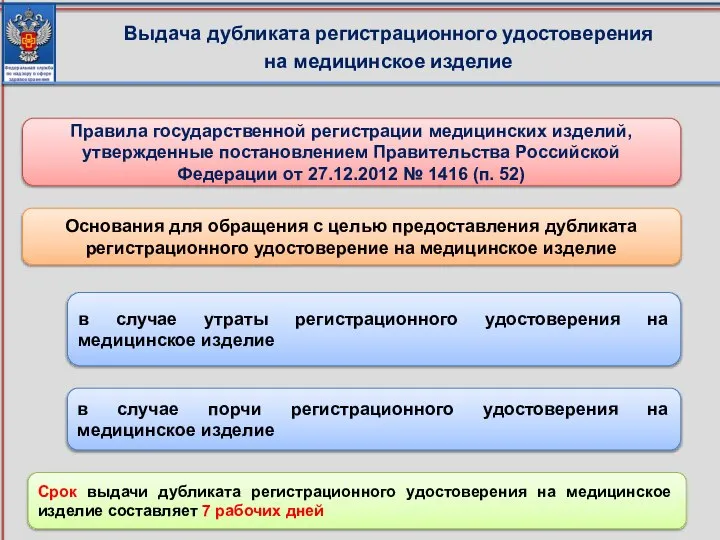
Слайд 29Выдача дубликата регистрационного удостоверения
на медицинское изделие
Правила государственной регистрации медицинских изделий, утвержденные постановлением
Выдача дубликата регистрационного удостоверения
на медицинское изделие
Правила государственной регистрации медицинских изделий, утвержденные постановлением
Слайд 30Документы и сведения, предоставляемые для выдачи дубликата регистрационного удостоверения на медицинское изделие
Заявление
Документы и сведения, предоставляемые для выдачи дубликата регистрационного удостоверения на медицинское изделие
Заявление

Слайд 31Доверенность от производителя медицинского изделия на выдачу дубликата регистрационного удостоверения с заверенным
Доверенность от производителя медицинского изделия на выдачу дубликата регистрационного удостоверения с заверенным

Слайд 32Часто встречающиеся основания для отказа в выдаче дубликата регистрационного удостоверения на медицинское
Часто встречающиеся основания для отказа в выдаче дубликата регистрационного удостоверения на медицинское


Слайд 34Федеральная служба по надзору в сфере здравоохранения
Основные нарушения и недостатки при рассмотрении
Федеральная служба по надзору в сфере здравоохранения
Основные нарушения и недостатки при рассмотрении

Слайд 35Нормативно-правовые акты, регламентирующие процедуры регистрации медицинских изделий и внесения изменений в регистрационные
Нормативно-правовые акты, регламентирующие процедуры регистрации медицинских изделий и внесения изменений в регистрационные

Слайд 36Государственная пошлина, предусмотренная налоговым законодательством Российской Федерации за процедуру внесения изменений в
Государственная пошлина, предусмотренная налоговым законодательством Российской Федерации за процедуру внесения изменений в

Слайд 37Внесение изменений в регистрационную документацию
(п. 55 Правил)
В случае необходимости (по желанию заявителя)
Внесение изменений в регистрационную документацию
(п. 55 Правил)
В случае необходимости (по желанию заявителя)

Слайд 38Внесение изменений в регистрационную документацию
(п. 55 Правил)
В случае необходимости внесения изменений в
Внесение изменений в регистрационную документацию
(п. 55 Правил)
В случае необходимости внесения изменений в

Слайд 39Внесение изменений в регистрационную документацию
(п. 55 Правил)
Вне зависимости от причины внесения изменений
Внесение изменений в регистрационную документацию
(п. 55 Правил)
Вне зависимости от причины внесения изменений

Слайд 40Внесение изменений в регистрационную документацию
(п. 55 Правил)
Основаниями для вынесения экспертным учреждением заключения
Внесение изменений в регистрационную документацию
(п. 55 Правил)
Основаниями для вынесения экспертным учреждением заключения

Слайд 41В случае если внесение изменений в техническую и (или) эксплуатационную документацию касаются:
Росздравнадзор
В случае если внесение изменений в техническую и (или) эксплуатационную документацию касаются:
Росздравнадзор

Слайд 42Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил
1. Окончание
Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил
1. Окончание

Слайд 43Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил
3. Изменение
Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил
3. Изменение

Слайд 44Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил
5. Изменение
Документы, необходимые для внесения изменений в регистрационное досье по п.55 Правил
5. Изменение


Слайд 46Федеральная служба по надзору в сфере здравоохранения
Требования к технической
и эксплуатационной документации
Федеральная служба по надзору в сфере здравоохранения
Требования к технической и эксплуатационной документации

Слайд 47Необходимость предоставления технической и эксплуатационной документации
Необходимость предоставления технической и эксплуатационной документации при
Необходимость предоставления технической и эксплуатационной документации
Необходимость предоставления технической и эксплуатационной документации при

Слайд 48Необходимость предоставления технической и эксплуатационной документации в рамках п. 37 Правил государственной
Необходимость предоставления технической и эксплуатационной документации в рамках п. 37 Правил государственной

Слайд 49Необходимость предоставления технической и эксплуатационной документации в рамках п. 55 Правил государственной

Слайд 50Техническая документация производителя (изготовителя)
Требования к технической документации установлены в пункте 4 Правил
Техническая документация производителя (изготовителя)
Требования к технической документации установлены в пункте 4 Правил

Слайд 51Эксплуатационная документация производителя (изготовителя)
Требования к эксплуатационной документации установлены
в пункте 4 Правил
Эксплуатационная документация производителя (изготовителя)
Требования к эксплуатационной документации установлены в пункте 4 Правил

Слайд 52Возможные варианты изменений, требующие актуализации технической и эксплуатационной документации
Изменение наименования изделия
Сведения
Возможные варианты изменений, требующие актуализации технической и эксплуатационной документации
Изменение наименования изделия
Сведения

Слайд 53Основные несоответствия технической и эксплуатационной документации производителя (изготовителя)
Основные несоответствия
Изменилось функциональное назначение
Расширились модельные
Основные несоответствия технической и эксплуатационной документации производителя (изготовителя)
Основные несоответствия
Изменилось функциональное назначение
Расширились модельные

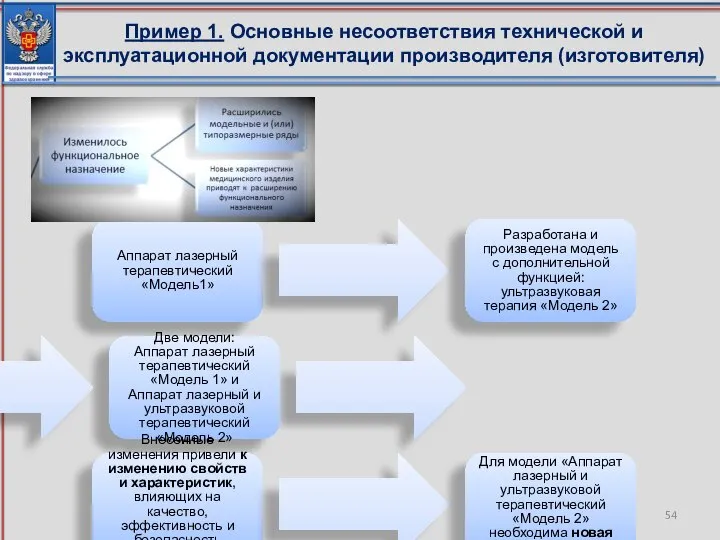
Слайд 54Пример 1. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя)
Аппарат лазерный терапевтический
Пример 1. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя)
Аппарат лазерный терапевтический
Слайд 55Пример 2. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя)
Изменены функциональные характеристики
Пример 2. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя)
Изменены функциональные характеристики

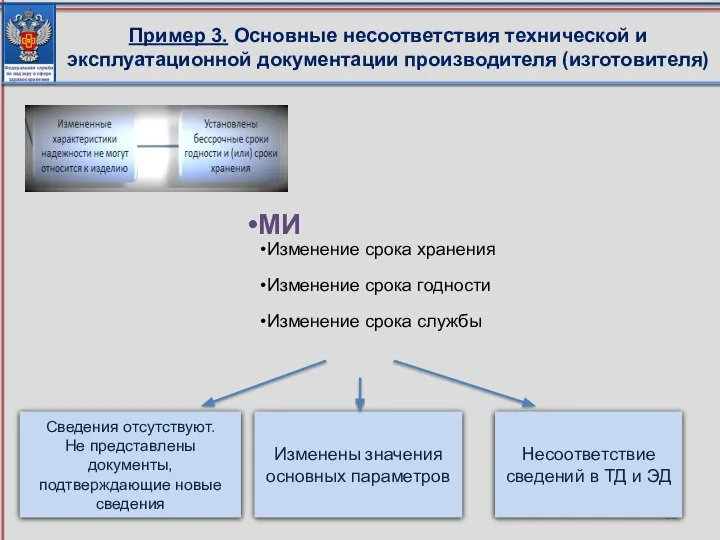
Слайд 56Пример 3. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя)
МИ
Изменение срока хранения
Изменение
Пример 3. Основные несоответствия технической и эксплуатационной документации производителя (изготовителя)
МИ
Изменение срока хранения
Изменение
Слайд 57Основные рекомендации для производителя при внесении изменений по п.55 Правил государственной регистрации
Основные рекомендации для производителя при внесении изменений по п.55 Правил государственной регистрации


Слайд 59Федеральная служба по надзору в сфере здравоохранения
Особенности оценки результатов технических испытаний
при
Федеральная служба по надзору в сфере здравоохранения
Особенности оценки результатов технических испытаний при

Слайд 60Пункт 55 - В случае необходимости (по желанию заявителя) внесения изменений в
Пункт 55 - В случае необходимости (по желанию заявителя) внесения изменений в

Слайд 61Пункт 4 Правил государственной регистрации медицинских изделий, утвержденных постановлением Правительства Российской Федерации
Пункт 4 Правил государственной регистрации медицинских изделий, утвержденных постановлением Правительства Российской Федерации

Слайд 62Оформление результатов технических испытаний для подтверждения внесенных изменений производится в соответствии с
Оформление результатов технических испытаний для подтверждения внесенных изменений производится в соответствии с

Слайд 63В п. 5 приказа Минздрава России от 09.01.2014 № 2н установлено:
«Технические испытания
В п. 5 приказа Минздрава России от 09.01.2014 № 2н установлено:
«Технические испытания

Слайд 64Порядок проведения технических испытаний медицинских изделий при внесении изменений
В рамках оценки и
Порядок проведения технических испытаний медицинских изделий при внесении изменений
В рамках оценки и

Слайд 65Перечень документов, которые предоставляет Заявитель для проведения технических испытаний (п. 9 приказа
Перечень документов, которые предоставляет Заявитель для проведения технических испытаний (п. 9 приказа

Слайд 66Акт результатов технических испытаний измененного медицинского изделия и Приложения к нему:
фотографическое изображение
фотографическое изображение

Слайд 67Перечень документов, подтверждающих результаты технических испытаний медицинских изделий
при внесении изменений
Акт
Программа технических испытаний
Фотографии
Протоколы
Перечень документов, подтверждающих результаты технических испытаний медицинских изделий
при внесении изменений
Акт
Программа технических испытаний
Фотографии
Протоколы

Слайд 68Перечень документов,
подтверждающих результаты технических испытаний медицинских изделий при внесении изменений
Акт оценки результатов
Перечень документов,
подтверждающих результаты технических испытаний медицинских изделий при внесении изменений
Акт оценки результатов

Слайд 69Структура Программы технических испытаний медицинских изделий при внесении изменений
- вводная часть;
- рассмотрение
Структура Программы технических испытаний медицинских изделий при внесении изменений
- вводная часть;
- рассмотрение

Слайд 70Структура и содержание протоколов испытаний (рекомендуемая)
ГОСТ ИСО/МЭК 17025-2009 Общие требования к компетентности
Структура и содержание протоколов испытаний (рекомендуемая)
ГОСТ ИСО/МЭК 17025-2009 Общие требования к компетентности

Слайд 71Структура и содержание протоколов испытаний (рекомендуемая)
- результаты проведенных испытаний медицинского изделия и
Структура и содержание протоколов испытаний (рекомендуемая)
- результаты проведенных испытаний медицинского изделия и

Слайд 72Распространенные замечания по результатам оценки протоколов технических испытаний внесенных изменений
Испытания проведены на
Распространенные замечания по результатам оценки протоколов технических испытаний внесенных изменений
Испытания проведены на

Слайд 73Рекомендации для проведения технических испытаний при внесении изменений в документацию
Проверить наличие действующей
Рекомендации для проведения технических испытаний при внесении изменений в документацию
Проверить наличие действующей


Слайд 75Федеральная служба по надзору в сфере здравоохранения
Особенности оценки результатов токсикологических исследований при
Федеральная служба по надзору в сфере здравоохранения
Особенности оценки результатов токсикологических исследований при

Слайд 76Пункт 4 Правил государственной регистрации медицинских изделий, утвержденных постановлением Правительства Российской Федерации
Пункт 4 Правил государственной регистрации медицинских изделий, утвержденных постановлением Правительства Российской Федерации

Слайд 77Оформление результатов токсикологических исследований для подтверждения внесенных изменений производится в соответствии с
Оформление результатов токсикологических исследований для подтверждения внесенных изменений производится в соответствии с

Слайд 78биологических в условиях in vitro и in vivo
санитарно-химических
Цель проведения токсикологических исследований медицинских
биологических в условиях in vitro и in vivo
санитарно-химических
Цель проведения токсикологических исследований медицинских
Слайд 79а) заявление о проведении токсикологических исследований;
б) образцы (образец) медицинского изделия или принадлежности,
а) заявление о проведении токсикологических исследований;
б) образцы (образец) медицинского изделия или принадлежности,

Слайд 80идентификация медицинского изделия (материала);
классификация медицинского изделия;
определение длительности контакта медицинского изделия
идентификация медицинского изделия (материала);
классификация медицинского изделия;
определение длительности контакта медицинского изделия

Слайд 81Медицинские изделия, в отношении которых проводятся токсикологические исследования
Согласно приказу Минздрава России от
Медицинские изделия, в отношении которых проводятся токсикологические исследования
Согласно приказу Минздрава России от
Слайд 82Медицинские изделия по виду контакта с организмом человека подразделяют на несколько групп.
Изделия,
Медицинские изделия по виду контакта с организмом человека подразделяют на несколько групп.
Изделия,

Слайд 83Программа токсикологических исследований составляется испытательной организацией совместно с заявителем и утверждается руководителем
Программа токсикологических исследований составляется испытательной организацией совместно с заявителем и утверждается руководителем

Слайд 84Соблюдение положений стандартов серии ISO 10993 "Оценка биологического действия медицинских изделий" позволит
Соблюдение положений стандартов серии ISO 10993 "Оценка биологического действия медицинских изделий" позволит

Слайд 851 этап: Санитарно-химические исследования
Санитарно-химические исследования позволяют отбраковывать непригодную к применению в клинической
1 этап: Санитарно-химические исследования
Санитарно-химические исследования позволяют отбраковывать непригодную к применению в клинической

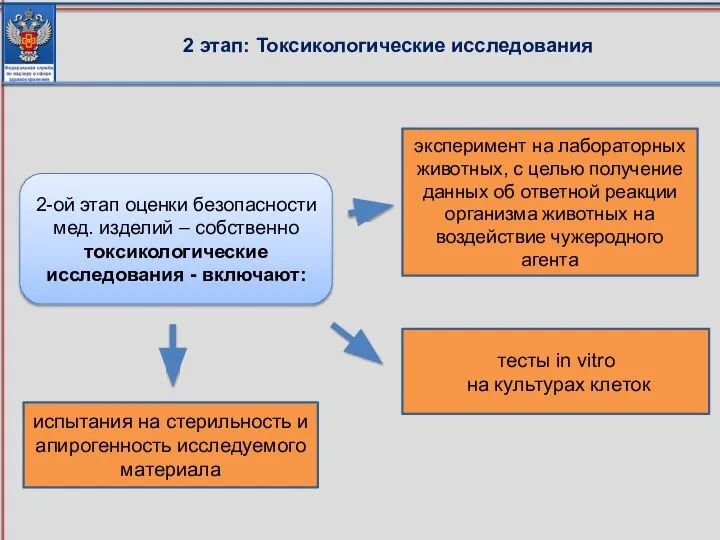
Слайд 862 этап: Токсикологические исследования
испытания на стерильность и апирогенность исследуемого материала
тесты in vitro
2 этап: Токсикологические исследования
испытания на стерильность и апирогенность исследуемого материала
тесты in vitro
Слайд 872 этап: Токсикологические исследования
- полноту и объективность установленных технической и эксплуатационной документацией
2 этап: Токсикологические исследования
- полноту и объективность установленных технической и эксплуатационной документацией

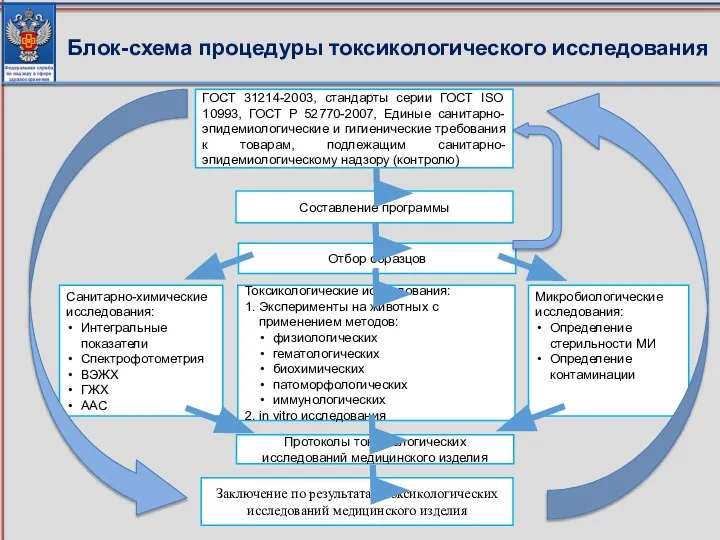
Слайд 88ГОСТ 31214-2003, стандарты серии ГОСТ ISO 10993, ГОСТ Р 52770-2007, Единые санитарно-эпидемиологические
ГОСТ 31214-2003, стандарты серии ГОСТ ISO 10993, ГОСТ Р 52770-2007, Единые санитарно-эпидемиологические
Слайд 89Оформляется заключение по результатам токсикологических исследований медицинского изделия, форма которого приведена в
Оформляется заключение по результатам токсикологических исследований медицинского изделия, форма которого приведена в

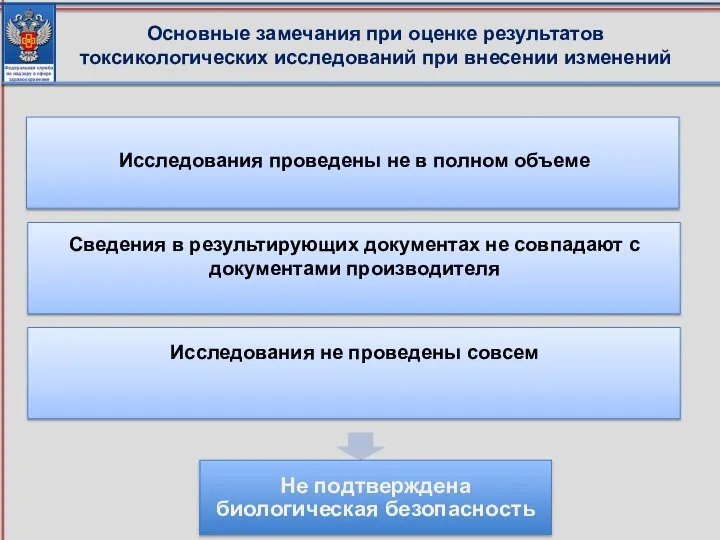
Слайд 90Исследования проведены не в полном объеме
Сведения в результирующих документах не совпадают с
Сведения в результирующих документах не совпадают с
Слайд 91Рекомендации для проведения токсикологических исследований при внесении изменений в документацию
Рекомендации для проведения токсикологических исследований при внесении изменений в документацию


Слайд 93Федеральная служба по надзору в сфере здравоохранения
Особенности оценки результатов клинических испытаний при
Федеральная служба по надзору в сфере здравоохранения
Особенности оценки результатов клинических испытаний при

Слайд 94Нормативно-правовое регулирование
при внесении изменений в регистрационное досье
Порядок проведения оценки соответствия медицинских
Нормативно-правовое регулирование
при внесении изменений в регистрационное досье
Порядок проведения оценки соответствия медицинских

Слайд 95Приложения и дополнения к Акту клинических испытаний
Следует обратить особое внимание, что
Приложения и дополнения к Акту клинических испытаний
Следует обратить особое внимание, что

Слайд 96Базы данных для поиска
EMBASE – Escerpta Medica published by Elsevier
CENTRAL – The
Базы данных для поиска EMBASE – Escerpta Medica published by Elsevier CENTRAL – The

Слайд 97Публикации о клинической эффективности
медицинского изделия
Публикации о клинической эффективности
медицинского изделия
Слайд 98Сведения о взаимозаменяемых медицинских изделиях
Сведения о взаимозаменяемых медицинских изделиях
Слайд 99Клинические испытания медицинских изделий
для диагностики in vitro
Клинические испытания медицинских изделий для
Клинические испытания медицинских изделий
для диагностики in vitro
Клинические испытания медицинских изделий для

Слайд 100Результаты клинических испытаний считаются отрицательными:
п. 45 Приказа Минздрава России от 09.01.2014
Результаты клинических испытаний считаются отрицательными:
п. 45 Приказа Минздрава России от 09.01.2014

Слайд 101Основания для проведения клинических испытаний
по п. 55 Правил государственной регистрации МИ
Расширение
Основания для проведения клинических испытаний
по п. 55 Правил государственной регистрации МИ
Расширение

Слайд 102Пример 1. Расширение показаний к применению
Изделие может использовать не только у взрослых,
Пример 1. Расширение показаний к применению
Изделие может использовать не только у взрослых,

Слайд 103Пример 2.
Уточнение/дополнение показаний к применению
При неизменности функционального назначения и (или) принципа
Пример 2.
Уточнение/дополнение показаний к применению
При неизменности функционального назначения и (или) принципа

Слайд 104Пример 3.
Уточнение/дополнение противопоказаний к применению
Противопоказания при регистрации МИ
- Введение в область
Пример 3.
Уточнение/дополнение противопоказаний к применению
Противопоказания при регистрации МИ - Введение в область

Слайд 105Пример 4.
Уточнение/дополнение побочных эффектов при применении
Новые побочные эффекты:
- уплотнение или узелки
Пример 4.
Уточнение/дополнение побочных эффектов при применении
Новые побочные эффекты:
- уплотнение или узелки

Слайд 106Пример 5. Расширение области применения
Области применения при регистрации:
Шейный отдел позвоночника
Запястье, кисть,
Пример 5. Расширение области применения
Области применения при регистрации:
Шейный отдел позвоночника
Запястье, кисть,

Слайд 107Пример 6. Модернизация режима работы
При неизменности функционального назначения и (или) принципа действия
Пример 6. Модернизация режима работы
При неизменности функционального назначения и (или) принципа действия

Слайд 108Пример 7. Добавление рабочей части и режима работы
При неизменности функционального назначения и
Пример 7. Добавление рабочей части и режима работы
При неизменности функционального назначения и
Слайд 109Пример 8. Расширение условий применения
При неизменности функционального назначения и (или) принципа действия
Пример 8. Расширение условий применения
При неизменности функционального назначения и (или) принципа действия

Слайд 110Пример 9. Совместимость медицинских изделий
При неизменности функционального назначения и (или) принципа действия
Пример 9. Совместимость медицинских изделий
При неизменности функционального назначения и (или) принципа действия
Слайд 111Пример 10. Уточнение/дополнение особых указаний при использовании МИ
При неизменности функционального назначения и
Пример 10. Уточнение/дополнение особых указаний при использовании МИ
При неизменности функционального назначения и
Слайд 112Федеральная служба по надзору в сфере здравоохранения
Анализ запросов о предоставлении дополнительных материалов
Федеральная служба по надзору в сфере здравоохранения
Анализ запросов о предоставлении дополнительных материалов

Слайд 113п. 26: При внесении изменений в документы, указанные в подпунктах "в" и

Слайд 114В случае недостаточности для вынесения экспертом заключения материалов и сведений, содержащихся в

Слайд 115Основаниями для вынесения экспертным учреждением заключения о невозможности внесения изменений в документы,

Слайд 116Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий
Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий
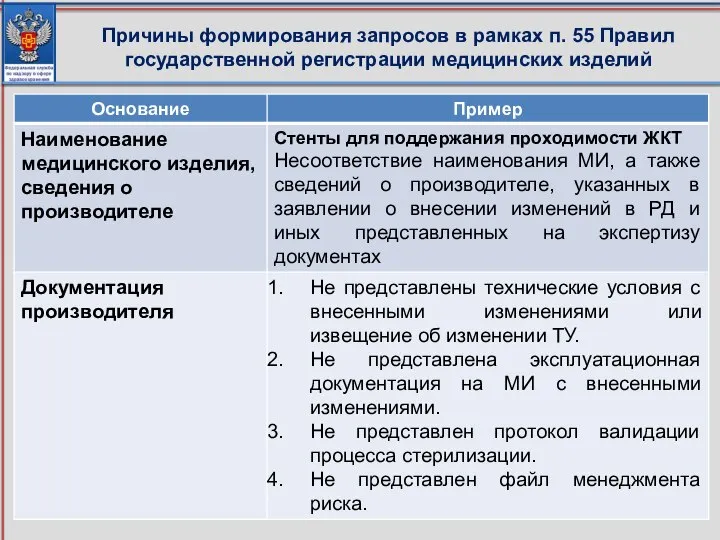
Слайд 117Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий
Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий

Слайд 118Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий
Причины формирования запросов в рамках п. 55 Правил государственной регистрации медицинских изделий

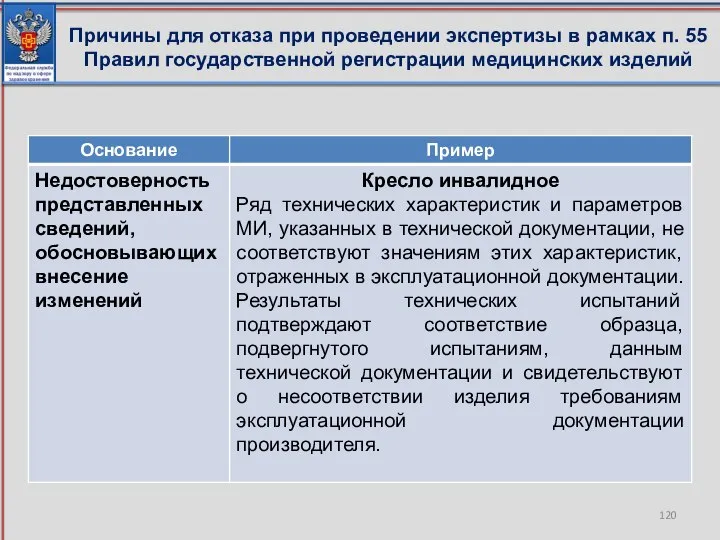
Слайд 119Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной

Слайд 120Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Слайд 121Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной

Слайд 122Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной

Слайд 123Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
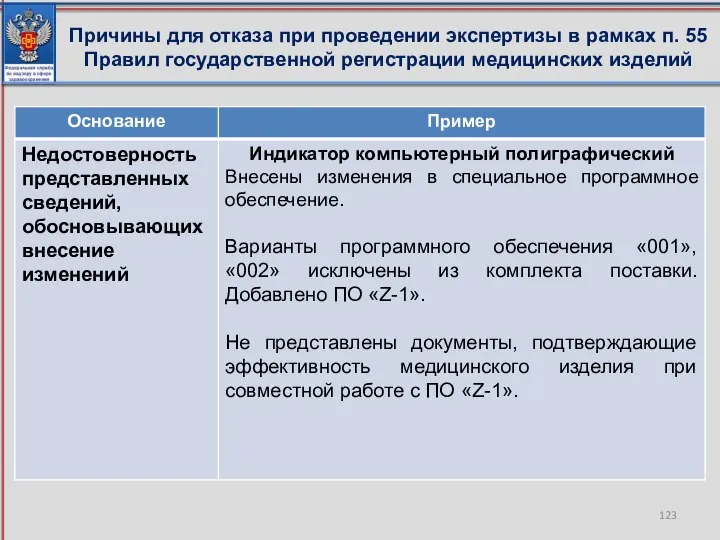
Слайд 124Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной

Слайд 125Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной

Слайд 126Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной

Слайд 127Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
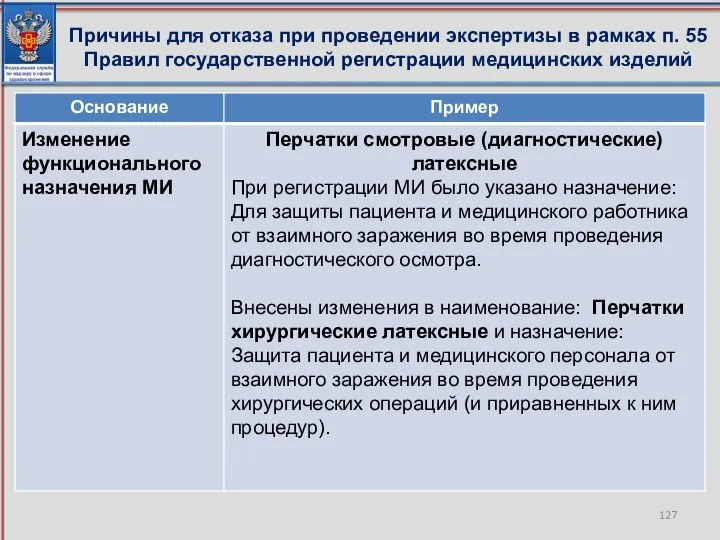
Слайд 128Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной

Слайд 129Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной
Причины для отказа при проведении экспертизы в рамках п. 55 Правил государственной


 Судебный этикет как составляющая культуры уголовно-процессуальной деятельности. Нормативные основы судебного этикета
Судебный этикет как составляющая культуры уголовно-процессуальной деятельности. Нормативные основы судебного этикета Оружие массового поражения
Оружие массового поражения Модельный ряд грузовых автомобилей Mercedes-Benz
Модельный ряд грузовых автомобилей Mercedes-Benz Энергетика: вчера, сегодня, завтра
Энергетика: вчера, сегодня, завтра ГИИС ЭБ. Запрос на аннулирование
ГИИС ЭБ. Запрос на аннулирование Элементы интернет-маркетинга и их взаимодействие
Элементы интернет-маркетинга и их взаимодействие 1. ОСНОВНЫЕ ПОНЯТИЯ Компьютерный исполнитель – это виртуальный объект, действующий в виртуальной среде обитания. Примеры: –Чертеж
1. ОСНОВНЫЕ ПОНЯТИЯ Компьютерный исполнитель – это виртуальный объект, действующий в виртуальной среде обитания. Примеры: –Чертеж Beerfest
Beerfest Красота осени
Красота осени Социально-психологическая служба в школе
Социально-психологическая служба в школе Как принимать управленческие решения
Как принимать управленческие решения Денежные переводы в Республике Таджикистан
Денежные переводы в Республике Таджикистан Темы в Drupal 6 Что нового, и чем оно грозит
Темы в Drupal 6 Что нового, и чем оно грозит Память в камне
Память в камне Помещение для открытия магазина Белорусская косметика
Помещение для открытия магазина Белорусская косметика Огневая подготовка. Версия 3
Огневая подготовка. Версия 3 Горный Дагестан
Горный Дагестан Методические рекомендации по введению модульной системы и системы зачетных единиц Методические рекомендации по разработке рабо
Методические рекомендации по введению модульной системы и системы зачетных единиц Методические рекомендации по разработке рабо Внутренний мир болезни
Внутренний мир болезни Техническое обслуживание и ремонт трансформатора ТДТНГ 40500\115\38,5\6,6 кВ Смоленск – 1
Техническое обслуживание и ремонт трансформатора ТДТНГ 40500\115\38,5\6,6 кВ Смоленск – 1 Кинестетик
Кинестетик Илья Ефимович Репин –великий русский художник
Илья Ефимович Репин –великий русский художник Václavské náměstí
Václavské náměstí Презентация на тему Иван третий (4 класс)
Презентация на тему Иван третий (4 класс) 11 причин инвестировать в Свердловскую область
11 причин инвестировать в Свердловскую область Технология установки врезного замка
Технология установки врезного замка Религия и мораль
Религия и мораль Караевское сельское поселение. Золотые руки мастера по бисероплетению
Караевское сельское поселение. Золотые руки мастера по бисероплетению