- пороки разв уха детей

Содержание
- 2. АКТУАЛЬНОСТЬ Врожденные пороки развития наружного и среднего уха в детском возрасте встречаются довольно часто. В среднем
- 3. Этиология. Пороки могут быть наследственными, но часто их причиной становится неблагоприятная экологическая обстановка, влияющая на беременную.
- 4. Общие подходы к лечению. Вопрос о хирургическом лечении этих больных зависит от состояния слуховой функции у
- 5. Рис. 2.38. Микротия. Рис. 2.37. Макротия и лопоухость.
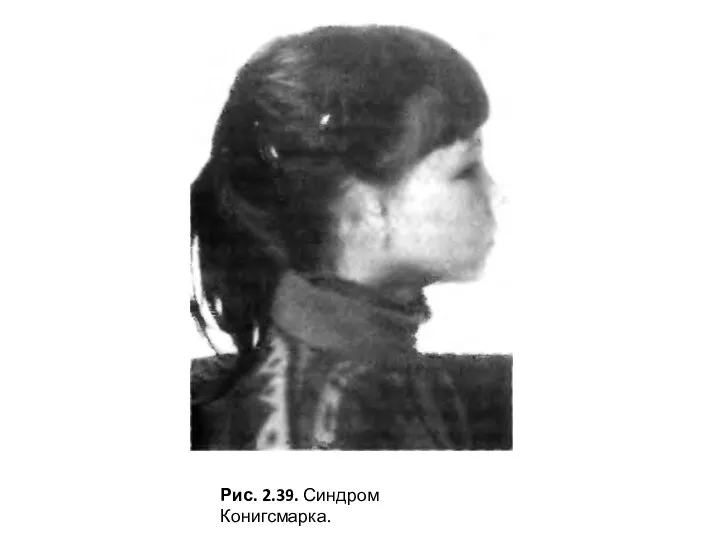
- 6. СИНДРОМ КОНИГСМАРКА Этиология. Синдром наследуется по аутосомно-рецессивному типу, шансы рождения второго ребенка с таким же пороком
- 7. Рис. 2.39. Синдром Конигсмарка.
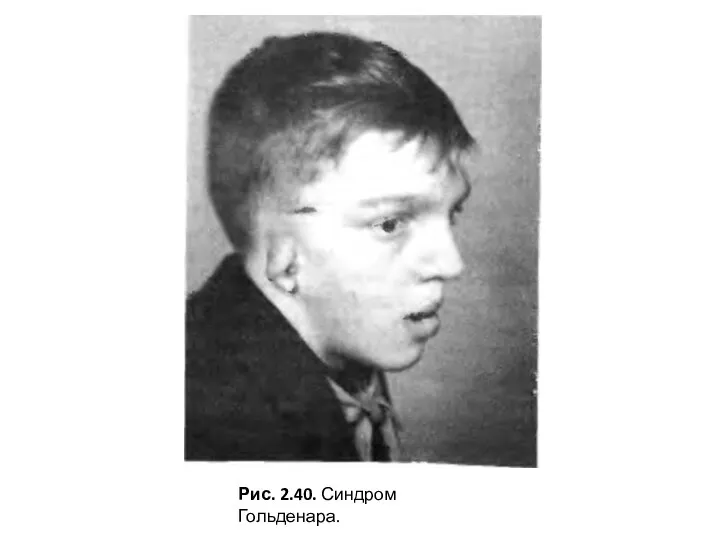
- 8. СИНДРОМ ГОЛЬДЕНАРА Синдром Гольденара — окулоаури-куловертебральная дисплазия. В литературе можно встретить и другое название — гемифациальная
- 9. Рис. 2.40. Синдром Гольденара.
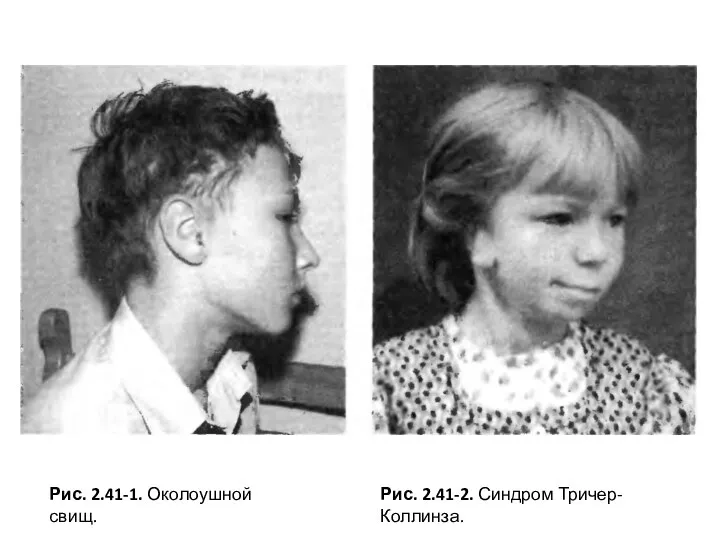
- 10. СИНДРОМ ТРИЧЕР-КОЛЛИНЗА (ФРАНЧЕСКЕТТИ) Этиология. Синдром наследуется по доминантному типу. Клинические проявления. У больных весьма характерное лицо,
- 11. Рис. 2.41-1. Околоушной свищ. Рис. 2.41-2. Синдром Тричер-Коллинза.
- 13. ВРОЖДЕННАЯ ПРЕАУРИКУЛЯРНАЯ ФИСТУЛА (ОКОЛОУШНОЙ СВИЩ) Этиология. В 25% случаев имеет наследственную природу, передаваясь по рецессивному типу.
- 15. Скачать презентацию
Слайд 2АКТУАЛЬНОСТЬ
Врожденные пороки развития наружного и среднего уха в детском возрасте встречаются довольно
АКТУАЛЬНОСТЬ
Врожденные пороки развития наружного и среднего уха в детском возрасте встречаются довольно

Слайд 3Этиология.
Пороки могут быть наследственными, но часто их причиной становится неблагоприятная экологическая обстановка,
Этиология.
Пороки могут быть наследственными, но часто их причиной становится неблагоприятная экологическая обстановка,

Слайд 4Общие подходы к лечению.
Вопрос о хирургическом лечении этих больных зависит от состояния
Общие подходы к лечению.
Вопрос о хирургическом лечении этих больных зависит от состояния

Слайд 5Рис. 2.38. Микротия.
Рис. 2.37. Макротия и лопоухость.
Рис. 2.38. Микротия.
Рис. 2.37. Макротия и лопоухость.

Слайд 6СИНДРОМ КОНИГСМАРКА
Этиология. Синдром наследуется по аутосомно-рецессивному типу, шансы рождения второго ребенка
СИНДРОМ КОНИГСМАРКА
Этиология. Синдром наследуется по аутосомно-рецессивному типу, шансы рождения второго ребенка

Слайд 7Рис. 2.39. Синдром Конигсмарка.
Рис. 2.39. Синдром Конигсмарка.
Слайд 8СИНДРОМ ГОЛЬДЕНАРА
Синдром Гольденара — окулоаури-куловертебральная дисплазия. В литературе можно встретить и
СИНДРОМ ГОЛЬДЕНАРА
Синдром Гольденара — окулоаури-куловертебральная дисплазия. В литературе можно встретить и

Слайд 9Рис. 2.40. Синдром Гольденара.
Рис. 2.40. Синдром Гольденара.
Слайд 10СИНДРОМ ТРИЧЕР-КОЛЛИНЗА (ФРАНЧЕСКЕТТИ)
Этиология. Синдром наследуется по доминантному типу.
Клинические проявления. У
СИНДРОМ ТРИЧЕР-КОЛЛИНЗА (ФРАНЧЕСКЕТТИ)
Этиология. Синдром наследуется по доминантному типу.
Клинические проявления. У

Слайд 11Рис. 2.41-1. Околоушной свищ.
Рис. 2.41-2. Синдром Тричер-Коллинза.
Рис. 2.41-1. Околоушной свищ.
Рис. 2.41-2. Синдром Тричер-Коллинза.

Слайд 13ВРОЖДЕННАЯ ПРЕАУРИКУЛЯРНАЯ ФИСТУЛА (ОКОЛОУШНОЙ СВИЩ)
Этиология. В 25% случаев имеет наследственную природу,
ВРОЖДЕННАЯ ПРЕАУРИКУЛЯРНАЯ ФИСТУЛА (ОКОЛОУШНОЙ СВИЩ)
Этиология. В 25% случаев имеет наследственную природу,

 ДНИ ВОИНСКОЙ СЛАВЫ РОССИИ
ДНИ ВОИНСКОЙ СЛАВЫ РОССИИ Классификация ошибок
Классификация ошибок Новый год в Германии
Новый год в Германии Дефис между частями слова в наречиях
Дефис между частями слова в наречиях Презентация на тему Микеланджело Буонарроти
Презентация на тему Микеланджело Буонарроти  0002dd4c-5d0c5ba0
0002dd4c-5d0c5ba0 Основные направления моды в парикмахерском искусстве
Основные направления моды в парикмахерском искусстве Малые формы для Diamante орех
Малые формы для Diamante орех The birth of cinematography
The birth of cinematography www.omnigrade.com
www.omnigrade.com ФАЙЛОВАЯ СИСТЕМА
ФАЙЛОВАЯ СИСТЕМА Я живу
Я живу Совещание руководителей муниципальных общеобразовательных учреждений Одинцовского муниципального района 11.11.11
Совещание руководителей муниципальных общеобразовательных учреждений Одинцовского муниципального района 11.11.11 Dnevnik.ru
Dnevnik.ru Федеральная служба по надзору в сфере образования и науки РФ. Задания для проведения ЕГЭ в компьютерной форм
Федеральная служба по надзору в сфере образования и науки РФ. Задания для проведения ЕГЭ в компьютерной форм Общая характеристика графического редактора
Общая характеристика графического редактора Исследовательская деятельность учителя
Исследовательская деятельность учителя Чередование звуков в корне и суффиксах. Е и О- беглые гласные
Чередование звуков в корне и суффиксах. Е и О- беглые гласные Работа установки ПРОЗА-2М в осеннем сеансе 2005 г.
Работа установки ПРОЗА-2М в осеннем сеансе 2005 г. Памятка для учащихся и их родителей. Что нужно знать о переводных экзаменах в 6-х, 7-х, 8-х, 10-х классах
Памятка для учащихся и их родителей. Что нужно знать о переводных экзаменах в 6-х, 7-х, 8-х, 10-х классах Отчет о выполнении работ по благоустройству общественной территории. Муниципальное образование Вельский муниципальный район
Отчет о выполнении работ по благоустройству общественной территории. Муниципальное образование Вельский муниципальный район Санкт-Петербург
Санкт-Петербург Презентация на тему Начало Гражданской войны в России
Презентация на тему Начало Гражданской войны в России  Криминалистическое исследование следов ног человека
Криминалистическое исследование следов ног человека Мезоэкономика: организация производства и менеджмент инноваций Заведующий кафедрой управления инновациями и организации произв
Мезоэкономика: организация производства и менеджмент инноваций Заведующий кафедрой управления инновациями и организации произв Презентация на тему История развития железнодорожного транспорта
Презентация на тему История развития железнодорожного транспорта  Органические вещества клетки. Белки
Органические вещества клетки. Белки Презентация на тему Федеральное агентство по рыболовству
Презентация на тему Федеральное агентство по рыболовству