- Синдром альпорта

Содержание
- 2. 25.12.2010 Синдром Альпорта Историческая справка: Первое упоминание патологии, известной как синдром Альпорта, принадлежит L. Guthrie, который
- 3. 25.12.2010 Синдром Альпорта Генетика: Гены: Col IV AIII Col IV AIV Col IV AV Col IV
- 4. 25.12.2010 Синдром Альпорта COL4A5 COL4A3-4
- 5. 25.12.2010 Синдром Альпорта Генетика: Число и тип мутаций: Коллаген IV типа состоит из трех доменов, плотно
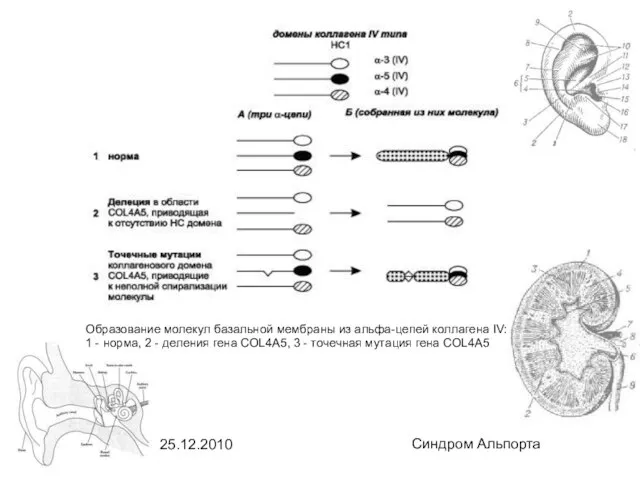
- 6. 25.12.2010 Синдром Альпорта Образование молекул базальной мембраны из альфа-цепей коллагена IV: 1 - норма, 2 -
- 7. 25.12.2010 Синдром Альпорта Белок: Состав:Коллаген 4 типа Состав Col IV AIII - 1260 аминокислотных остатков Col
- 8. 25.12.2010 Синдром Альпорта Белок: Структурные особенности: Молекула коллагена представляет собой правозакрученную спираль из трёх α-цепей. Один
- 9. 25.12.2010 Синдром Альпорта
- 10. 25.12.2010 Синдром Альпорта Белок: Функциональность: Белок(молекулы коллагена IV типа) обеспечивает нерастворимость и механическую стабильность базальных мембран,
- 11. 25.12.2010 Синдром Альпорта Признаки болезни в организме: Основные симптомы: Клиническая картина синдрома Альпорта, регулярно повторяющаяся в
- 12. 25.12.2010 Синдром Альпорта Признаки болезни в организме: Связь с генетическими заболеваниями: При болезни Шарко-Мари-Тута семейное сочетание
- 13. 25.12.2010 Синдром Альпорта Признаки болезни в организме: Типы пораженных клеток: Нейроны, волосяные клетки Необычные особенности болезни:
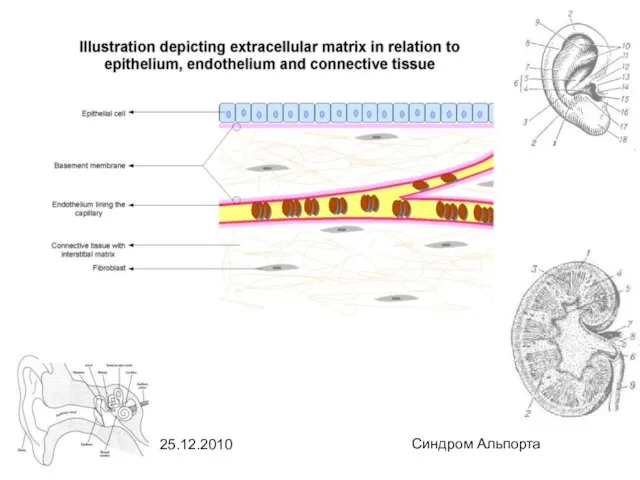
- 14. 25.12.2010 Синдром Альпорта Клеточная и молекулярная биология: Поражение органелл: Поражаются базальные мембраны. Базальная мембрана - это
- 15. 25.12.2010 Синдром Альпорта Клеточная и молекулярная биология: Нормальные функции: В нормально развивающейся почке, изначально коллаген α1
- 16. 25.12.2010 Синдром Альпорта Клеточная и молекулярная биология: Как мутации изменяют функции органеллы: Патология аллельных вариантов Подавляющее
- 18. Скачать презентацию
Слайд 225.12.2010
Синдром Альпорта
Историческая справка:
Первое упоминание патологии, известной как синдром Альпорта, принадлежит L.
25.12.2010
Синдром Альпорта
Историческая справка:
Первое упоминание патологии, известной как синдром Альпорта, принадлежит L.

Слайд 325.12.2010
Синдром Альпорта
Генетика:
Гены: Col IV AIII Col IV AIV Col IV AV Col
25.12.2010
Синдром Альпорта
Генетика:
Гены: Col IV AIII Col IV AIV Col IV AV Col

Слайд 425.12.2010
Синдром Альпорта
COL4A5 COL4A3-4
25.12.2010
Синдром Альпорта
COL4A5 COL4A3-4

Слайд 525.12.2010
Синдром Альпорта
Генетика:
Число и тип мутаций:
Коллаген IV типа состоит из трех доменов, плотно
25.12.2010
Синдром Альпорта
Генетика:
Число и тип мутаций:
Коллаген IV типа состоит из трех доменов, плотно

Слайд 625.12.2010
Синдром Альпорта
Образование молекул базальной мембраны из альфа-цепей коллагена IV: 1 - норма,
25.12.2010
Синдром Альпорта
Образование молекул базальной мембраны из альфа-цепей коллагена IV: 1 - норма,
Слайд 725.12.2010
Синдром Альпорта
Белок:
Состав:Коллаген 4 типа
Состав
Col IV AIII - 1260 аминокислотных остатков
Col IV AIV
25.12.2010
Синдром Альпорта
Белок:
Состав:Коллаген 4 типа
Состав
Col IV AIII - 1260 аминокислотных остатков
Col IV AIV

Слайд 825.12.2010
Синдром Альпорта
Белок:
Структурные особенности:
Молекула коллагена представляет собой правозакрученную спираль из трёх α-цепей. Один
25.12.2010
Синдром Альпорта
Белок:
Структурные особенности:
Молекула коллагена представляет собой правозакрученную спираль из трёх α-цепей. Один

Слайд 925.12.2010
Синдром Альпорта
25.12.2010
Синдром Альпорта
Слайд 1025.12.2010
Синдром Альпорта
Белок:
Функциональность:
Белок(молекулы коллагена IV типа) обеспечивает нерастворимость и механическую стабильность базальных мембран,
25.12.2010
Синдром Альпорта
Белок:
Функциональность:
Белок(молекулы коллагена IV типа) обеспечивает нерастворимость и механическую стабильность базальных мембран,

Слайд 1125.12.2010
Синдром Альпорта
Признаки болезни в организме:
Основные симптомы:
Клиническая картина синдрома Альпорта, регулярно повторяющаяся в
25.12.2010
Синдром Альпорта
Признаки болезни в организме:
Основные симптомы:
Клиническая картина синдрома Альпорта, регулярно повторяющаяся в

Слайд 1225.12.2010
Синдром Альпорта
Признаки болезни в организме:
Связь с генетическими заболеваниями:
При болезни Шарко-Мари-Тута семейное сочетание
25.12.2010
Синдром Альпорта
Признаки болезни в организме:
Связь с генетическими заболеваниями:
При болезни Шарко-Мари-Тута семейное сочетание

Слайд 1325.12.2010
Синдром Альпорта
Признаки болезни в организме:
Типы пораженных клеток:
Нейроны, волосяные клетки
Необычные особенности болезни:
Поражение
25.12.2010
Синдром Альпорта
Признаки болезни в организме:
Типы пораженных клеток:
Нейроны, волосяные клетки
Необычные особенности болезни:
Поражение

Слайд 1425.12.2010
Синдром Альпорта
Клеточная и молекулярная биология:
Поражение органелл:
Поражаются базальные мембраны.
Базальная мембрана - это плотное
25.12.2010
Синдром Альпорта
Клеточная и молекулярная биология:
Поражение органелл:
Поражаются базальные мембраны.
Базальная мембрана - это плотное

Слайд 1525.12.2010
Синдром Альпорта
Клеточная и молекулярная биология:
Нормальные функции:
В нормально развивающейся почке, изначально коллаген α1
25.12.2010
Синдром Альпорта
Клеточная и молекулярная биология:
Нормальные функции:
В нормально развивающейся почке, изначально коллаген α1

Слайд 1625.12.2010
Синдром Альпорта
Клеточная и молекулярная биология:
Как мутации изменяют функции органеллы:
Патология аллельных вариантов
Подавляющее большинство
25.12.2010
Синдром Альпорта
Клеточная и молекулярная биология:
Как мутации изменяют функции органеллы:
Патология аллельных вариантов
Подавляющее большинство

 Volleyball
Volleyball Овеянные славою флаг наш и герб
Овеянные славою флаг наш и герб Презентация на тему Разнообразие горных пород
Презентация на тему Разнообразие горных пород Скелет. Строение, состав и соединение костей
Скелет. Строение, состав и соединение костей Проект «Презентация : + и –»
Проект «Презентация : + и –» Определение каналов сбыта
Определение каналов сбыта Планирование маршрута доставки груза в смешанном сообщении
Планирование маршрута доставки груза в смешанном сообщении Презентация на тему Вещие сны
Презентация на тему Вещие сны Типология обществ
Типология обществ  Прибор для определения вероятности изменения погоды
Прибор для определения вероятности изменения погоды Презентация на тему к уроку по рассказу А.П.Чехова Хамелеон
Презентация на тему к уроку по рассказу А.П.Чехова Хамелеон  Реклама на современном этапе
Реклама на современном этапе Противоатеросклеротические средства.
Противоатеросклеротические средства. Эффективность работы системы дополнительного образования с целью повышения качества образования.
Эффективность работы системы дополнительного образования с целью повышения качества образования. Презентация на тему Логика и ее свойства
Презентация на тему Логика и ее свойства Философия Древней Греции
Философия Древней Греции Презентация на тему Имидж
Презентация на тему Имидж  УкрКульт 1921-1953
УкрКульт 1921-1953 Как древние люди представляли себе Вселенную
Как древние люди представляли себе Вселенную Яндекс
Яндекс Структурная типология языков
Структурная типология языков Традиции Бурятии
Традиции Бурятии Система управления эффективностью бизнеса и персонала
Система управления эффективностью бизнеса и персонала Логотип Разработка фирменного стиля Визитка Папка.
Логотип Разработка фирменного стиля Визитка Папка. Специальные средства ОВД
Специальные средства ОВД Защита теоретической части выпускной квалификационной работы
Защита теоретической части выпускной квалификационной работы Анализ цикла документальных фильмов Девяностые
Анализ цикла документальных фильмов Девяностые Всемирное наследие
Всемирное наследие