- Генетика пола и наследственные нарушения половой дифференцировки

Содержание
- 2. Очевидно, что дифференцировка пола происходит на нескольких уровнях: 1) генетическом; 2) гонадном; 3) гормональном; 4) фенотипическом;
- 3. Этапы созревания половых клеток
- 4. У человека первичные половые клетки могут быть обнаружены в первичной полоске уже на 16-18 день развития,
- 5. В течение последующих 3-4 месяцев оогонии делятся митотически, в результате их количество от исходных 1500-2000 возрастает
- 6. Они окружаются фолликулярными клетками и в виде ооцитов 1-го порядка сохраняются до половой зрелости. Количество ооцитов
- 7. До момента овуляции выжившие ооциты I порядка проходят период роста и вновь вступают в мейоз, который
- 8. У половозрелых мужчин общая продолжительность сперматогенеза составляет 70-72 дня. За это время стволовые клетки сперматогенного ряда
- 9. Затем они окружаются клетками целомического эпителия, образуя «половые тяжи», и остаются в недифференцированном виде вплоть до
- 10. После митотического деления и дифференцировки сперматогонии вступают в два последовательных деления мейоза, образуя сперматоциты 1-го, а
- 11. Во время второй фазы сперматогенеза – эпидидемальной – завершается созревание спермиев, и они приобретают подвижность. Ежедневно
- 12. До 6-недельного возраста зачатки гонад у эмбриона развиваются как бипотенциальные образования. Их дифференцировка в семенник или
- 13. Развитие по мужскому типу и дифференцировка гонады в семенник запускается тестис-детерминирующим фактором (TDF), появление которого обусловлено
- 14. Генетический контроль и наследственные нарушения половой дифференцировки
- 15. Таким образом, определяющую роль в первичной детерминации пола у млекопитающих играет Y-хромосома. Это свойство Y-хромосомы определяется
- 16. При мужском кариотипе и делеции гена SRY развитие идет по женскому типу, хотя при этом оно
- 17. С другой стороны перенос на X-хромосому или аутосому локуса Yp11.31, содержащего ген SRY, приводит к формированию
- 18. В детерминации пола участвуют не только гены Y-хромосомы, но также аутосомные и Х-хромосомные гены. В настоящее
- 19. Среди первой группы SRY-родственный ген SOX9. Ко второй группе относится ген DAX1. В морфогенезе гонад на
- 20. До начала половой дифференцировки выводящая система эмбриона представлена вольфовыми и мюллеровыми протоками. Обособление наружных половых органов
- 21. У мужчин наоборот, мюллеровы протоки атрофируются, а вольфовы преобразуются в семявыносящие протоки и семенные пузырьки. За
- 22. Ген WNT4 экспрессируется в желтом теле и его продукт на ранних стадиях фолликулярного роста, участвует в
- 23. Половая дифференцировка гонад сама по себе недостаточна для формирования мужского или женского фенотипа. Вторичная детерминация пола
- 24. В формировании мужского фенотипа участвуют два гормона: антимюллеровый, вызывающий регрессию мюллеровых протоков, и тестостерон, или андроген,
- 25. Ген антимюллерового гормона AMH активируется геном SRY на 7-ой неделе беременности, и его экспрессия продолжается до
- 26. Важную роль в детерминации пола играют также эстрогены и их рецепторы. Однако при недостаточности мужских половых
- 27. Мы перечислили только ключевые этапы первичной половой дифференцировки. Функциональные взаимоотношения между контролирующими эти процессы генами, определяют
- 28. Наследственные нарушения мужской половой дифференцировки, или реверсии пола XY (РПXY) — это генетически гетерогенная группа заболеваний,
- 29. Одним из генетических вариантов РПXY является синдром Свайера. К сходному фенотипу могут приводить дупликации гена DAX1.
- 30. К РПXY могут приводить доминантные мутации в гене SF1 и микроделеции цитогенетической области 9p24.3. В последнем
- 31. Аутосомно-рецессивный тип РПXY обусловлен мутациями в гене транскрипционного фактора, участвующего в морфогенезе гонад на ранней бипотенциальной
- 32. Аутосомно-доминантные типы РПXY могут быть обусловлены мутациями в генах митоген-активирующей киназы, участвующей в контроле апоптоза (MAP3K1);
- 33. Реверсия пола при мужском кариотипе может наблюдаться у больных кампомелической дисплазией, обусловленной мутациями в гене транскрипционного
- 34. Поэтому неудивительно, что некоторые мутации в гене WT1 сопровождаются генитальными аномалиями у мужчин, вплоть до полной
- 35. Причиной реверсии пола ХХ (РПХХ), то есть развития половой системы по мужскому типу при кариотипе 46,XX,
- 36. Мутации в гене WNT4 обнаруживаются при двух аллельных заболеваниях с РПХХ: аутосомно-доминантной аплазии мюллеровых протоков у
- 37. Некоторые мутации в перечисленных выше генах приводят к различным формам гермафродитизма. К мужскому ложному гермафродитизму могут
- 38. Больные мужчины при нормальном кариотипе имеют женское строение наружных половых органов в сочетании с гипоспадией и
- 39. Дефекты метаболизма тестостерона в периферических тканях, обусловленные мутациями в гене стероидной 5-альфа-редуктазы 2 (SRD5A2), также являются
- 40. В большинстве случаев наружные половые органы имеют женский тип при наличии яичек в паховом канале. Иногда
- 41. Врожденный идиопатический гипогонадотропный гипогонадизм (ВИГГ) характеризуется низким уровнем циркулирующих гонадотропинов и тестостерона с выраженной задержкой полового
- 42. Эта патология может развиваться при недостаточной секреции гонадотропин-рилизинг гормона (ГнРГ) – декапептида гипотоламуса, который является ключевым
- 43. В настоящее время идентифицированы гены 18 наследственных типов ВИГГ. Эти гены составляют генетическую сеть, ответственную за
- 44. Почти у 20% больных ВИГГ одновременно присутствуют мутации в двух и более генах, причем в большинстве
- 45. Генетически гетерогенный синдром Каллмана объясняет около 20% всех случаев ВИГГ. Х-сцепленный тип заболевания обусловлен мутациями в
- 46. При аутосомно-доминантном типе мутации найдены в гене рецептора 1 фибробластных факторов роста (FGFR1) иногда в сочетании
- 47. Вторыми по частоте являются типы ВИГГ, обусловленные мутациями в генах прокинецитина 2 (PROK2) и его рецептора
- 48. Прокинецитины – это небольшие секреторные белки, участвующие в регуляции циркадных ритмов. Они присутствуют в гипоталамусе и
- 49. Мужское и женское бесплодие
- 50. Наследственные причины первичного бесплодия достаточно разнообразны, при этом нужно различать мужское и женское бесплодие. Все варианты
- 51. Нарушения гаметогенеза являются одним из ведущих проявлений хромосомных болезней. У 2-3% мужчин выявляется олигозооспермия или азооспермия.
- 52. До 30% случаев необструктивной азооспермии и тяжелой олигозооспермии вызваны микроделециями, возникающими de novo в клетках сперматогенного
- 53. Чаще всего эти дефекты располагаются в дистальной части длинного плеча Y-хромосомы, где находится локус AZF, получивший
- 54. Этот локус содержит 3 участка (AZFa, AZFb и AZFc), в каждом из которых присутствуют гены, участвующие
- 55. В среднем, частота микроделеций в локусе AZF при азооспермии составляет 15% и при тяжелой олигозооспермии –
- 56. Конечно, AZF – не единственный локус, связанный со сперматогенезом. Наследственная сперматогенная недостаточность (СН) может быть связана
- 57. К первому классу относится СН, обусловленная мутациями в гене белка 3 синаптонемального комплекса (SYCP3). Причиной другого
- 58. Необычная морфология сперматозоидов (крупная головка, многожгутиковость) в сочетании с их полиплоидией, обусловлена мутациями в гене AURKC,
- 59. Однако в 75% случаев причиной глобозооспермии у мужчин разного этнического происхождения является присутствие гомозиготных 200-кб делеций,
- 60. При муковисцидозе наблюдается прогрессирующая непроходимость семявыводящих протоков и нарушения сперматогенеза. Больные муковисцидозом мужчины, как правило, бесплодны
- 61. В настоящее время описаны более 80 мутаций в гене муковисцидоза (CFTR), которые приводят к врожденному билатеральному
- 62. Кроме того, частота гетерозиготных носителей мутаций в гене CFTR среди пациентов с билатеральным отсутствием семявыносящих протоков
- 63. Наследственными вариантами первичного женского бесплодия являются гипергонадотропная дисгенезия яичников и синдром преждевременной недостаточности яичников. Дизгенезия яичников
- 64. Аутосомно-рецессивная гипергонадотропная дисгенезия яичников 1-го типа в сочетании с тяжелым остеопорозом и некоторыми другими нарушениями обусловлена
- 65. Причиной развития Х-сцепленной дизгенезии яичников 2-го типа являются доминантные мутации в гене костного морфогенетического белка, участвующего
- 66. Синдром преждевременной недостаточности яичников – это гетерогенная группа заболеваний, при которых вторичная аменорея с повышенным уровнем
- 67. Три из них оказались локализованы в длинном плече Х-хромосомы – FMR1, DIAPH2 и POF1В. Мутации в
- 68. Напомним, что динамические мутации в гене FMR1, обусловленные экспансией CGG-повтора, приводят к синдрому Мартина-Белл. У больных
- 69. Ген DIAPH2 участвует в контроле цитокинеза, а ген POF1В кодирует актин-связывающий белок. В некоторых случаях у
- 70. Еще один сцепленный с Х-хромосомой доминантный тип преждевременной недостаточности яичников является аллельным вариантом дизгенезии яичников 2
- 71. Адрено-генитальный синдром
- 72. Адреногенитальный синдром (АГС) — это гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями в генах ферментов биосинтеза стероидных
- 73. При этом нарушается выработка кортизола надпочечниками, и его дефицит приводит к избыточной продукции АКТГ-зависимых стероидов. Общая
- 75. Наиболее распространенным является 3 тип АГС, или синдром дефицита 21-гидроксилазы, объясняющий более 90% всех случаев заболевания.
- 76. Следствием дефицита 21-гидроксилазы является уменьшение содержания кортизола и альдостерона в плазме крови, избыточная секреция АКТГ, повышение
- 77. Накопление 17-гидроксипрогестерона вызывает потерю натрия и задержку калия, что сопровождается потерей воды. Гиперэкспрессия АКТГ стимулирует синтез
- 78. Заболевание характеризуется значительным клиническим полиморфизмом. Классические варианты делятся на тяжелую сольтеряющую и более легкую, простую (вирильную)
- 79. В первом случае у больных наблюдаются выраженные нарушения солевого обмена в виде гипонатриемии и гиперкалиемии, которые
- 80. Длительность кризов варьирует от нескольких минут до получаса, а иногда они заканчиваются гибелью больного. На 2–3-й
- 81. Нарастание электролитных нарушений приводит к развитию гипонатриемической дегидратации, гиперкалиемии, метаболическому ацидозу и кардиогенному шоку. Нарушения солевого
- 82. Простая форма заболевания, составляющая 1/3 всех случаев, проявляется в виде изолированной вирилизации наружных половых органов без
- 83. У мальчиков вирильная форма диагностируется с 5 лет и старше при появлении признаков преждевременного полового и
- 84. Неклассические или взрослые варианты АГС включают различные нарушения полового созревания в подростковом или пубертатном возрасте в
- 85. Ген CYP21A2 расположен в области локализации генов HLA-комплекса, которая отличается высокой рекомбиногенной и мутагенной активностью. В
- 86. Спектр мутаций в гене CYP21A2 у больных АГС-3 хорошо изучен. Широкое распространение в разных популяциях имеют
- 87. При классических вариантах АГС3 – сольтеряющей и простой – выявляются разные мажорные мутации. В первом случае
- 89. Среди пациентов с подозрением на неклассический вариант врожденной гиперплазии коры надпочечников мутации в гене CYP21A2 идентифицируются
- 91. Синдром тестикулярной феминизации
- 92. Синдром тестикулярной феминизации, или синдром нечувствительности к андрогенам — Х-сцепленное рецессивное заболевание, обусловленное присутствием инактивирующих мутаций
- 93. Патогенез синдрома связан с полным или частичным отсутствием чувствительности тканей к андрогенам. Распространенность заболевания составляет 1
- 94. В зависимости от степени нечувствительности периферических рецепторов к андрогенам различают полную форму и неполную форму, когда
- 95. Клинически болезнь у мальчиков проявляется в виде ложного мужского гермафродитизма при нормальном мужском кариотипе 46,XY. Полная
- 96. Половые железы представлены яичками, которые располагаются внутрибрюшинно, в половых губах или паховом канале, формируя грыжевое выпячивание.
- 97. Молочные железы хорошо развиты, при этом подмышечное и лобковое оволосение — скудные. Телосложение, эмоциональное и сексуальное
- 98. Неполные формы синдрома тестикулярной феминизации обусловлены снижением количества рецепторов на поверхности клеток-мишеней или их связывающей способности
- 99. Фенотип больных, как правило, мужской, но сопровождается различной степенью гипоплазии половых органов, крипторхизмом, гипоспадией, расщеплением мошонки
- 100. Спектр мутаций в гене AR достаточно разнообразен. При полной форме синдрома тестикулярной феминизации у больных обнаруживаются
- 101. Целью лечения больных с полной формой синдрома тестикулярной феминизации является предотвращение опухолевого перерождения тестикул (удаление половых
- 102. При неполной форме необходимо предотвратить пубертатную вирилизацию наружных половых органов и огрубение голоса. В послеоперационном периоде
- 103. Интересно отметить, что в первом экзоне гена AR локализован нестабильный тринуклеотидный CAG-повтор, экспансия которого, сопровождающаяся увеличением
- 105. Скачать презентацию
Слайд 2Очевидно, что дифференцировка пола происходит на нескольких уровнях: 1) генетическом;
2) гонадном;
Очевидно, что дифференцировка пола происходит на нескольких уровнях: 1) генетическом; 2) гонадном;

Слайд 3Этапы созревания половых клеток
Этапы созревания половых клеток

Слайд 4У человека первичные половые клетки могут быть обнаружены в первичной полоске уже
У человека первичные половые клетки могут быть обнаружены в первичной полоске уже

Слайд 5В течение последующих 3-4 месяцев оогонии делятся митотически, в результате их количество
В течение последующих 3-4 месяцев оогонии делятся митотически, в результате их количество

Слайд 6Они окружаются фолликулярными клетками и в виде ооцитов 1-го порядка сохраняются до
Они окружаются фолликулярными клетками и в виде ооцитов 1-го порядка сохраняются до

Слайд 7До момента овуляции выжившие ооциты I порядка проходят период роста и вновь
До момента овуляции выжившие ооциты I порядка проходят период роста и вновь

Слайд 8У половозрелых мужчин общая продолжительность сперматогенеза составляет 70-72 дня.
За это время
У половозрелых мужчин общая продолжительность сперматогенеза составляет 70-72 дня. За это время

Слайд 9Затем они окружаются клетками целомического эпителия, образуя «половые тяжи», и остаются в
Затем они окружаются клетками целомического эпителия, образуя «половые тяжи», и остаются в

Слайд 10После митотического деления и дифференцировки сперматогонии вступают в два последовательных деления мейоза,
После митотического деления и дифференцировки сперматогонии вступают в два последовательных деления мейоза,

Слайд 11Во время второй фазы сперматогенеза – эпидидемальной – завершается созревание спермиев, и
Во время второй фазы сперматогенеза – эпидидемальной – завершается созревание спермиев, и

Слайд 12До 6-недельного возраста зачатки гонад у эмбриона развиваются как бипотенциальные образования.
Их
До 6-недельного возраста зачатки гонад у эмбриона развиваются как бипотенциальные образования. Их

Слайд 13Развитие по мужскому типу и дифференцировка гонады в семенник запускается тестис-детерминирующим фактором
Развитие по мужскому типу и дифференцировка гонады в семенник запускается тестис-детерминирующим фактором

Слайд 14Генетический контроль и наследственные нарушения половой дифференцировки
Генетический контроль и наследственные нарушения половой дифференцировки

Слайд 15Таким образом, определяющую роль в первичной детерминации пола у млекопитающих играет Y-хромосома.
Таким образом, определяющую роль в первичной детерминации пола у млекопитающих играет Y-хромосома.

Слайд 16При мужском кариотипе и делеции гена SRY развитие идет по женскому типу,
При мужском кариотипе и делеции гена SRY развитие идет по женскому типу,

Слайд 17С другой стороны перенос на X-хромосому или аутосому локуса Yp11.31, содержащего ген
С другой стороны перенос на X-хромосому или аутосому локуса Yp11.31, содержащего ген

Слайд 18В детерминации пола участвуют не только гены Y-хромосомы, но также аутосомные и
В детерминации пола участвуют не только гены Y-хромосомы, но также аутосомные и

Слайд 19Среди первой группы SRY-родственный ген SOX9. Ко второй группе относится ген DAX1.
Среди первой группы SRY-родственный ген SOX9. Ко второй группе относится ген DAX1.

Слайд 20До начала половой дифференцировки выводящая система эмбриона представлена вольфовыми и мюллеровыми протоками.
До начала половой дифференцировки выводящая система эмбриона представлена вольфовыми и мюллеровыми протоками.

Слайд 21У мужчин наоборот, мюллеровы протоки атрофируются, а вольфовы преобразуются в семявыносящие протоки
У мужчин наоборот, мюллеровы протоки атрофируются, а вольфовы преобразуются в семявыносящие протоки

Слайд 22Ген WNT4 экспрессируется в желтом теле и его продукт на ранних стадиях
Ген WNT4 экспрессируется в желтом теле и его продукт на ранних стадиях

Слайд 23Половая дифференцировка гонад сама по себе недостаточна для формирования мужского или женского
Половая дифференцировка гонад сама по себе недостаточна для формирования мужского или женского

Слайд 24В формировании мужского фенотипа участвуют два гормона: антимюллеровый, вызывающий регрессию мюллеровых протоков,
В формировании мужского фенотипа участвуют два гормона: антимюллеровый, вызывающий регрессию мюллеровых протоков,

Слайд 25Ген антимюллерового гормона AMH активируется геном SRY на 7-ой неделе беременности, и
Ген антимюллерового гормона AMH активируется геном SRY на 7-ой неделе беременности, и

Слайд 26Важную роль в детерминации пола играют также эстрогены и их рецепторы.
Однако
Важную роль в детерминации пола играют также эстрогены и их рецепторы. Однако

Слайд 27Мы перечислили только ключевые этапы первичной половой дифференцировки. Функциональные взаимоотношения между контролирующими
Мы перечислили только ключевые этапы первичной половой дифференцировки. Функциональные взаимоотношения между контролирующими

Слайд 28Наследственные нарушения мужской половой дифференцировки, или реверсии пола XY (РПXY) — это
Наследственные нарушения мужской половой дифференцировки, или реверсии пола XY (РПXY) — это

Слайд 29Одним из генетических вариантов РПXY является синдром Свайера.
К сходному фенотипу могут приводить
Одним из генетических вариантов РПXY является синдром Свайера. К сходному фенотипу могут приводить

Слайд 30К РПXY могут приводить доминантные мутации в гене SF1 и микроделеции цитогенетической
К РПXY могут приводить доминантные мутации в гене SF1 и микроделеции цитогенетической

Слайд 31Аутосомно-рецессивный тип РПXY обусловлен мутациями в гене транскрипционного фактора, участвующего в морфогенезе
Аутосомно-рецессивный тип РПXY обусловлен мутациями в гене транскрипционного фактора, участвующего в морфогенезе

Слайд 32Аутосомно-доминантные типы РПXY могут быть обусловлены мутациями в генах митоген-активирующей киназы, участвующей
Аутосомно-доминантные типы РПXY могут быть обусловлены мутациями в генах митоген-активирующей киназы, участвующей

Слайд 33Реверсия пола при мужском кариотипе может наблюдаться у больных кампомелической дисплазией, обусловленной
Реверсия пола при мужском кариотипе может наблюдаться у больных кампомелической дисплазией, обусловленной

Слайд 34Поэтому неудивительно, что некоторые мутации в гене WT1 сопровождаются генитальными аномалиями у
Поэтому неудивительно, что некоторые мутации в гене WT1 сопровождаются генитальными аномалиями у

Слайд 35Причиной реверсии пола ХХ (РПХХ), то есть развития половой системы по мужскому
Причиной реверсии пола ХХ (РПХХ), то есть развития половой системы по мужскому

Слайд 36Мутации в гене WNT4 обнаруживаются при двух аллельных заболеваниях с РПХХ: аутосомно-доминантной
Мутации в гене WNT4 обнаруживаются при двух аллельных заболеваниях с РПХХ: аутосомно-доминантной

Слайд 37Некоторые мутации в перечисленных выше генах приводят к различным формам гермафродитизма. К
Некоторые мутации в перечисленных выше генах приводят к различным формам гермафродитизма. К

Слайд 38Больные мужчины при нормальном кариотипе имеют женское строение наружных половых органов в
Больные мужчины при нормальном кариотипе имеют женское строение наружных половых органов в

Слайд 39Дефекты метаболизма тестостерона в периферических тканях, обусловленные мутациями в гене стероидной 5-альфа-редуктазы
Дефекты метаболизма тестостерона в периферических тканях, обусловленные мутациями в гене стероидной 5-альфа-редуктазы

Слайд 40В большинстве случаев наружные половые органы имеют женский тип при наличии яичек
В большинстве случаев наружные половые органы имеют женский тип при наличии яичек

Слайд 41Врожденный идиопатический гипогонадотропный гипогонадизм (ВИГГ) характеризуется низким уровнем циркулирующих гонадотропинов и тестостерона
Врожденный идиопатический гипогонадотропный гипогонадизм (ВИГГ) характеризуется низким уровнем циркулирующих гонадотропинов и тестостерона

Слайд 42Эта патология может развиваться при недостаточной секреции гонадотропин-рилизинг гормона (ГнРГ) – декапептида
Эта патология может развиваться при недостаточной секреции гонадотропин-рилизинг гормона (ГнРГ) – декапептида

Слайд 43В настоящее время идентифицированы гены 18 наследственных типов ВИГГ.
Эти гены составляют
В настоящее время идентифицированы гены 18 наследственных типов ВИГГ. Эти гены составляют

Слайд 44Почти у 20% больных ВИГГ одновременно присутствуют мутации в двух и более
Почти у 20% больных ВИГГ одновременно присутствуют мутации в двух и более

Слайд 45Генетически гетерогенный синдром Каллмана объясняет около 20% всех случаев ВИГГ.
Х-сцепленный тип
Генетически гетерогенный синдром Каллмана объясняет около 20% всех случаев ВИГГ. Х-сцепленный тип

Слайд 46При аутосомно-доминантном типе мутации найдены в гене рецептора 1 фибробластных факторов роста
При аутосомно-доминантном типе мутации найдены в гене рецептора 1 фибробластных факторов роста

Слайд 47Вторыми по частоте являются типы ВИГГ, обусловленные мутациями в генах прокинецитина 2
Вторыми по частоте являются типы ВИГГ, обусловленные мутациями в генах прокинецитина 2

Слайд 48Прокинецитины – это небольшие секреторные белки, участвующие в регуляции циркадных ритмов.
Они
Прокинецитины – это небольшие секреторные белки, участвующие в регуляции циркадных ритмов. Они

Слайд 49Мужское и женское бесплодие
Мужское и женское бесплодие

Слайд 50Наследственные причины первичного бесплодия достаточно разнообразны, при этом нужно различать мужское и
Наследственные причины первичного бесплодия достаточно разнообразны, при этом нужно различать мужское и

Слайд 51Нарушения гаметогенеза являются одним из ведущих проявлений хромосомных болезней.
У 2-3% мужчин
Нарушения гаметогенеза являются одним из ведущих проявлений хромосомных болезней. У 2-3% мужчин

Слайд 52До 30% случаев необструктивной азооспермии и тяжелой олигозооспермии вызваны микроделециями, возникающими de
До 30% случаев необструктивной азооспермии и тяжелой олигозооспермии вызваны микроделециями, возникающими de

Слайд 53 Чаще всего эти дефекты располагаются в дистальной части длинного плеча Y-хромосомы,
Чаще всего эти дефекты располагаются в дистальной части длинного плеча Y-хромосомы,

Слайд 54Этот локус содержит 3 участка
(AZFa, AZFb и AZFc),
в каждом из
Этот локус содержит 3 участка (AZFa, AZFb и AZFc), в каждом из

Слайд 55В среднем, частота микроделеций в локусе AZF при азооспермии составляет 15% и
В среднем, частота микроделеций в локусе AZF при азооспермии составляет 15% и

Слайд 56Конечно, AZF – не единственный локус, связанный со сперматогенезом.
Наследственная
сперматогенная недостаточность
Конечно, AZF – не единственный локус, связанный со сперматогенезом. Наследственная сперматогенная недостаточность

Слайд 57К первому классу относится СН, обусловленная мутациями в гене белка 3 синаптонемального
К первому классу относится СН, обусловленная мутациями в гене белка 3 синаптонемального

Слайд 58Необычная морфология сперматозоидов (крупная головка, многожгутиковость) в сочетании с их полиплоидией, обусловлена
Необычная морфология сперматозоидов (крупная головка, многожгутиковость) в сочетании с их полиплоидией, обусловлена

Слайд 59Однако в 75% случаев причиной глобозооспермии у мужчин разного этнического происхождения является
Однако в 75% случаев причиной глобозооспермии у мужчин разного этнического происхождения является

Слайд 60При муковисцидозе наблюдается прогрессирующая непроходимость семявыводящих протоков и нарушения сперматогенеза.
Больные муковисцидозом
При муковисцидозе наблюдается прогрессирующая непроходимость семявыводящих протоков и нарушения сперматогенеза. Больные муковисцидозом

Слайд 61В настоящее время описаны более 80 мутаций в гене муковисцидоза (CFTR), которые
В настоящее время описаны более 80 мутаций в гене муковисцидоза (CFTR), которые

Слайд 62Кроме того, частота гетерозиготных носителей мутаций в гене CFTR среди пациентов с
Кроме того, частота гетерозиготных носителей мутаций в гене CFTR среди пациентов с

Слайд 63Наследственными вариантами первичного женского бесплодия являются гипергонадотропная дисгенезия яичников и синдром преждевременной
Наследственными вариантами первичного женского бесплодия являются гипергонадотропная дисгенезия яичников и синдром преждевременной

Слайд 64Аутосомно-рецессивная гипергонадотропная дисгенезия яичников 1-го типа в сочетании с тяжелым остеопорозом и
Аутосомно-рецессивная гипергонадотропная дисгенезия яичников 1-го типа в сочетании с тяжелым остеопорозом и

Слайд 65Причиной развития
Х-сцепленной дизгенезии яичников 2-го типа являются доминантные мутации в гене
Причиной развития Х-сцепленной дизгенезии яичников 2-го типа являются доминантные мутации в гене

Слайд 66Синдром преждевременной недостаточности яичников – это гетерогенная группа заболеваний, при которых вторичная
Синдром преждевременной недостаточности яичников – это гетерогенная группа заболеваний, при которых вторичная

Слайд 67Три из них оказались локализованы в длинном плече Х-хромосомы – FMR1, DIAPH2
Три из них оказались локализованы в длинном плече Х-хромосомы – FMR1, DIAPH2

Слайд 68Напомним, что динамические мутации в гене FMR1, обусловленные экспансией CGG-повтора, приводят к
Напомним, что динамические мутации в гене FMR1, обусловленные экспансией CGG-повтора, приводят к

Слайд 69Ген DIAPH2 участвует в контроле цитокинеза, а ген POF1В кодирует актин-связывающий белок.
Ген DIAPH2 участвует в контроле цитокинеза, а ген POF1В кодирует актин-связывающий белок.

Слайд 70Еще один сцепленный с Х-хромосомой доминантный тип преждевременной недостаточности яичников является аллельным
Еще один сцепленный с Х-хромосомой доминантный тип преждевременной недостаточности яичников является аллельным

Слайд 71Адрено-генитальный синдром
Адрено-генитальный синдром

Слайд 72Адреногенитальный синдром (АГС) — это гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями в
Адреногенитальный синдром (АГС) — это гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями в

Слайд 73При этом нарушается выработка кортизола надпочечниками, и его дефицит приводит к избыточной
При этом нарушается выработка кортизола надпочечниками, и его дефицит приводит к избыточной

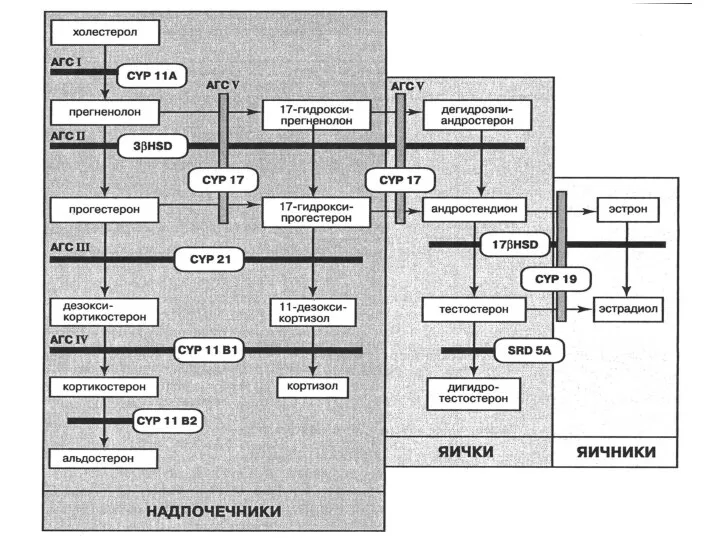
Слайд 75Наиболее распространенным является 3 тип АГС, или синдром дефицита 21-гидроксилазы, объясняющий более
Наиболее распространенным является 3 тип АГС, или синдром дефицита 21-гидроксилазы, объясняющий более

Слайд 76Следствием дефицита 21-гидроксилазы является уменьшение содержания кортизола и альдостерона в плазме крови,
Следствием дефицита 21-гидроксилазы является уменьшение содержания кортизола и альдостерона в плазме крови,

Слайд 77Накопление 17-гидроксипрогестерона вызывает потерю натрия и задержку калия, что сопровождается потерей воды.
Накопление 17-гидроксипрогестерона вызывает потерю натрия и задержку калия, что сопровождается потерей воды.

Слайд 78Заболевание характеризуется значительным клиническим полиморфизмом. Классические варианты делятся на тяжелую сольтеряющую и
Заболевание характеризуется значительным клиническим полиморфизмом. Классические варианты делятся на тяжелую сольтеряющую и

Слайд 79В первом случае у больных наблюдаются выраженные нарушения солевого обмена в виде
В первом случае у больных наблюдаются выраженные нарушения солевого обмена в виде

Слайд 80Длительность кризов варьирует от нескольких минут до получаса, а иногда они заканчиваются
Длительность кризов варьирует от нескольких минут до получаса, а иногда они заканчиваются

Слайд 81Нарастание электролитных нарушений приводит к развитию гипонатриемической дегидратации, гиперкалиемии, метаболическому ацидозу и
Нарастание электролитных нарушений приводит к развитию гипонатриемической дегидратации, гиперкалиемии, метаболическому ацидозу и

Слайд 82Простая форма заболевания, составляющая 1/3 всех случаев, проявляется в виде изолированной вирилизации
Простая форма заболевания, составляющая 1/3 всех случаев, проявляется в виде изолированной вирилизации

Слайд 83У мальчиков вирильная форма диагностируется с 5 лет и старше при появлении
У мальчиков вирильная форма диагностируется с 5 лет и старше при появлении

Слайд 84Неклассические или взрослые варианты АГС включают различные нарушения полового созревания в подростковом
Неклассические или взрослые варианты АГС включают различные нарушения полового созревания в подростковом

Слайд 85Ген CYP21A2 расположен в области локализации генов HLA-комплекса, которая отличается высокой рекомбиногенной
Ген CYP21A2 расположен в области локализации генов HLA-комплекса, которая отличается высокой рекомбиногенной

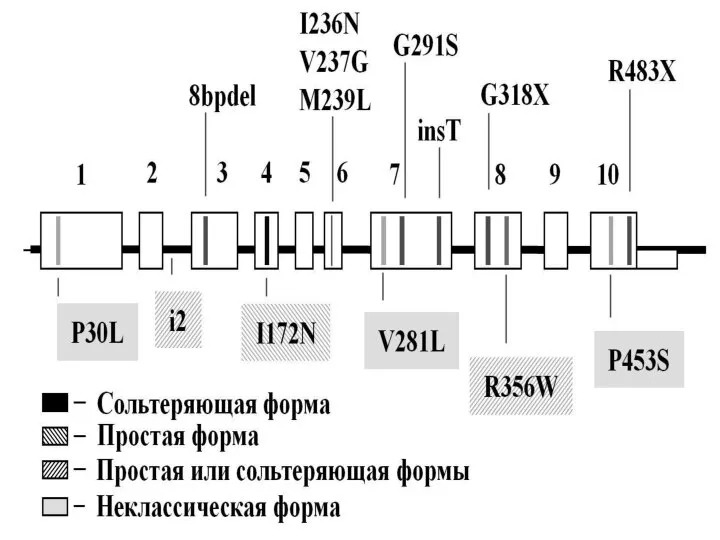
Слайд 86Спектр мутаций в гене CYP21A2 у больных АГС-3 хорошо изучен. Широкое распространение
Спектр мутаций в гене CYP21A2 у больных АГС-3 хорошо изучен. Широкое распространение

Слайд 87При классических вариантах АГС3 – сольтеряющей и простой – выявляются разные мажорные
При классических вариантах АГС3 – сольтеряющей и простой – выявляются разные мажорные


Слайд 89Среди пациентов с подозрением на неклассический вариант врожденной гиперплазии коры надпочечников мутации
Среди пациентов с подозрением на неклассический вариант врожденной гиперплазии коры надпочечников мутации

Слайд 91Синдром тестикулярной феминизации
Синдром тестикулярной феминизации

Слайд 92Синдром тестикулярной феминизации, или синдром нечувствительности к андрогенам — Х-сцепленное рецессивное заболевание,
Синдром тестикулярной феминизации, или синдром нечувствительности к андрогенам — Х-сцепленное рецессивное заболевание,

Слайд 93Патогенез синдрома связан с полным или частичным отсутствием чувствительности тканей к андрогенам.
Патогенез синдрома связан с полным или частичным отсутствием чувствительности тканей к андрогенам.

Слайд 94В зависимости от степени нечувствительности периферических рецепторов к андрогенам различают полную форму
В зависимости от степени нечувствительности периферических рецепторов к андрогенам различают полную форму

Слайд 95Клинически болезнь у мальчиков проявляется в виде ложного мужского гермафродитизма при нормальном
Клинически болезнь у мальчиков проявляется в виде ложного мужского гермафродитизма при нормальном

Слайд 96Половые железы представлены яичками, которые располагаются внутрибрюшинно, в половых губах или паховом
Половые железы представлены яичками, которые располагаются внутрибрюшинно, в половых губах или паховом

Слайд 97Молочные железы хорошо развиты, при этом подмышечное и лобковое оволосение — скудные.
Молочные железы хорошо развиты, при этом подмышечное и лобковое оволосение — скудные.

Слайд 98Неполные формы синдрома тестикулярной феминизации обусловлены снижением количества рецепторов на поверхности клеток-мишеней
Неполные формы синдрома тестикулярной феминизации обусловлены снижением количества рецепторов на поверхности клеток-мишеней

Слайд 99Фенотип больных, как правило, мужской, но сопровождается различной степенью гипоплазии половых органов,
Фенотип больных, как правило, мужской, но сопровождается различной степенью гипоплазии половых органов,

Слайд 100Спектр мутаций в гене AR достаточно разнообразен. При полной форме синдрома тестикулярной
Спектр мутаций в гене AR достаточно разнообразен. При полной форме синдрома тестикулярной

Слайд 101Целью лечения больных с полной формой синдрома тестикулярной феминизации является предотвращение опухолевого
Целью лечения больных с полной формой синдрома тестикулярной феминизации является предотвращение опухолевого

Слайд 102При неполной форме необходимо предотвратить пубертатную вирилизацию наружных половых органов и огрубение
При неполной форме необходимо предотвратить пубертатную вирилизацию наружных половых органов и огрубение

Слайд 103Интересно отметить, что в первом экзоне гена AR локализован нестабильный тринуклеотидный CAG-повтор,
Интересно отметить, что в первом экзоне гена AR локализован нестабильный тринуклеотидный CAG-повтор,

 Витамин PP
Витамин PP Рыбы глубоководной пелагиали
Рыбы глубоководной пелагиали Лекция. Печень, поджелудочная железа (2)
Лекция. Печень, поджелудочная железа (2) Надежная защита организма
Надежная защита организма Геннің құрлымы
Геннің құрлымы Сравнение Голосеменных и Покрытосеменных растений
Сравнение Голосеменных и Покрытосеменных растений Биосфера. Условия существования организмов
Биосфера. Условия существования организмов Спинной мозг
Спинной мозг Презентация на тему СТРОЕНИЕ ПТИЦ
Презентация на тему СТРОЕНИЕ ПТИЦ  Тип Моллюски
Тип Моллюски Современная ззология
Современная ззология Значение пчел в жизни человека
Значение пчел в жизни человека Химический состав клетки
Химический состав клетки Лесовосстановление путем внесения в почву дополнительных веществ
Лесовосстановление путем внесения в почву дополнительных веществ Цепи питания
Цепи питания Как паук костюмчик менял
Как паук костюмчик менял Способности человека
Способности человека Эндокринная система
Эндокринная система Лабораторная работа №3 Изучение способов поглощения пищи у животных (4)
Лабораторная работа №3 Изучение способов поглощения пищи у животных (4) Тема урока: Строение цветка
Тема урока: Строение цветка Деление клетки. Клеточный цикл
Деление клетки. Клеточный цикл Основные этапы развития органического мира Земли
Основные этапы развития органического мира Земли Спинной мозг, его строение и функции
Спинной мозг, его строение и функции Презентация на тему Мозг
Презентация на тему Мозг  Пища. Питательные вещества и природные пищевые компоненты – важный экологический фактор
Пища. Питательные вещества и природные пищевые компоненты – важный экологический фактор Химия ферментов
Химия ферментов Приспособления 9 кл
Приспособления 9 кл Витамины и здоровье
Витамины и здоровье