- Молекулярная биология

Содержание
- 2. Некоторые параметры молекул ДНК и белка: Один шаг это полный виток спирали ДНК-поворот на 3600 Один
- 3. Выравнивание генетических последовательностей В эволюции генетических последовательностей происходят как замены, так и вставки и делеции. Первым
- 4. Выравнивание генетических последовательностей
- 5. Выравнивание генетических последовательностей
- 6. Выравнивание генетических последовательностей Clustal -- это одна из самых широко используемых компьютерных программ для множественного выравнивания
- 7. Выбираем нужный тип данных (белок, ДНК или РНК) В нашем случае - DNA
- 8. 2. Вставляем последовательности Открываем папку Выравнивания. Открываем файл В-11.fasta программой UltraEdit-32. Копируем оттуда 2 или более
- 9. 3. Нажимаем Submit
- 10. 4. Получаем результат
- 11. 5. Интерпретация Звездочка (*) – различия по данной позиции (нуклеотид или аминокислота) отсутствуют между разными последовательностями
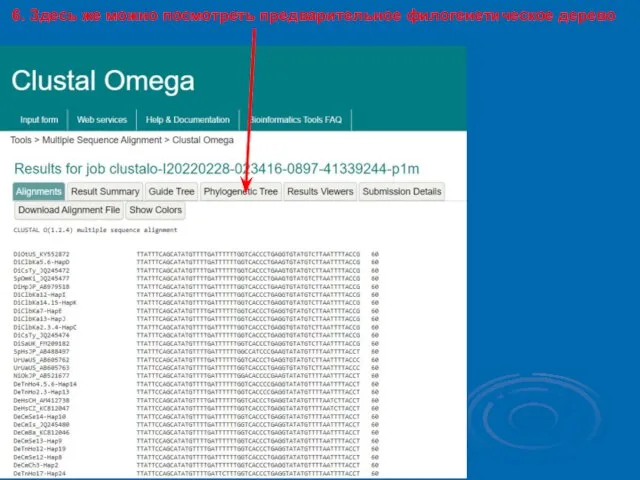
- 12. 6. Здесь же можно посмотреть предварительное филогенетическое дерево
- 14. BLAST BLAST (англ. Basic Local Alignment Search Tool — средство поиска основного локального выравнивания) — семейство
- 15. Классификация программ серии BLAST Нуклеотидные предназначены для сравнения изучаемой нуклеотидной последовательности с базой данных секвенированных нуклеиновых
- 16. Классификация программ серии BLAST Белковые предназначены для сравнения изучаемой аминокислотной последовательности белка с имеющейся базой данных
- 17. Классификация программ серии BLAST Транслирующие способны транслировать нуклеотидные последовательности в аминокислотные: blastx — переводит изучаемую нуклеотидную
- 18. Классификация программ серии BLAST Геномные предназначены для сравнения изучаемой нуклеотидной последовательности с базой данных секвенированного генома
- 19. Переходим в сервис BLAST Национального центра биотехнологической информации США (NCBI) по ссылке: https://blast.ncbi.nlm.nih.gov/Blast.cgi
- 20. blastn tblastn blastx blastp
- 21. Задача 1. Форма отчета Каждый лично на своем компьютере делает скриншот/фото (так, чтобы было видно номер
- 22. Задача 1 Открыть с помощью программы Chromas файл с хроматограммой Задание_1-R.20120413T.A11 из папки Задания Это последовательность
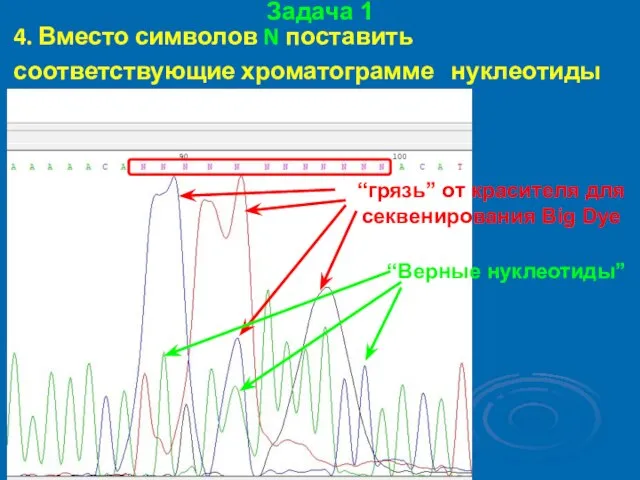
- 24. Задача 1 4. Вместо символов N поставить соответствующие хроматограмме нуклеотиды (A, T, G или С): “грязь”
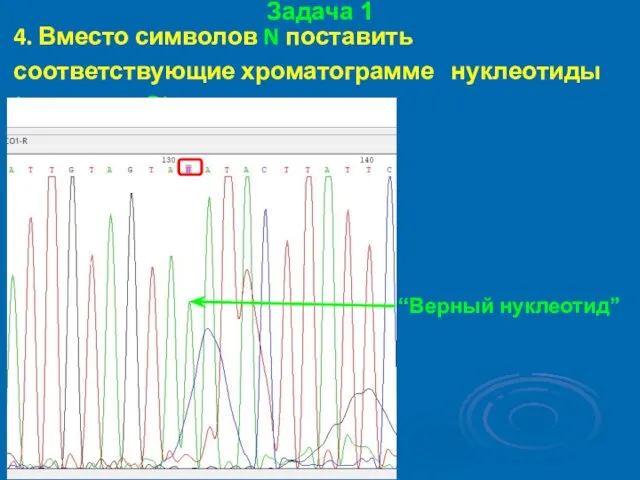
- 25. Задача 1 4. Вместо символов N поставить соответствующие хроматограмме нуклеотиды (A, T, G или С): “Верный
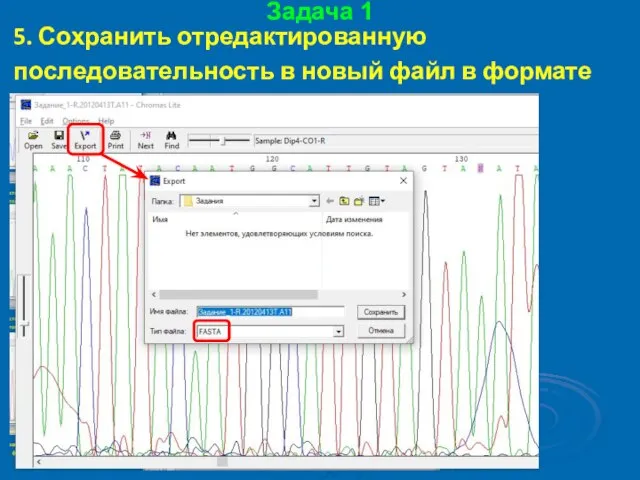
- 26. Задача 1 5. Сохранить отредактированную последовательность в новый файл в формате FASTA:
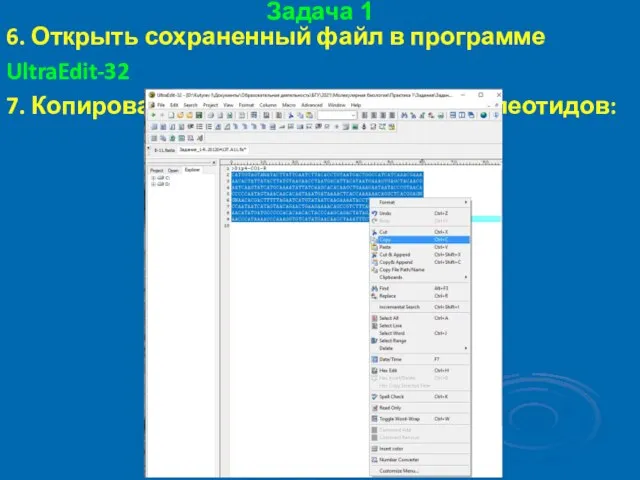
- 27. Задача 1 6. Открыть сохраненный файл в программе UltraEdit-32 7. Копировать последовательность нуклеотидов:
- 28. Задача 1 8. Переходим в сервис BLAST : https://blast.ncbi.nlm.nih.gov/Blast.cgi blastn
- 29. Задача 1 9. Вставляем последовательность в окошко 10. Нажимаем кнопку BLAST:
- 30. Задача 1 11. Получаем в итоге список гомологов, близких к нашей последовательности
- 31. Задача 1. Форма отчета Каждый лично на своем компьютере делает скриншот/фото (так, чтобы было видно номер
- 32. Задача 2. Форма отчета Каждый лично отправляет мне на почту [email protected] : 1) файл insulin.fasta 2)
- 33. Задача 2 Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
- 34. Задача 2 Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
- 35. Задача 2 Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
- 36. Задача 2 Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
- 37. Задача 2 2. С помощью программы blastn найти в нуклеотидном виде гомологи этой последовательности https://blast.ncbi.nlm.nih.gov/Blast.cgi
- 38. Задача 2 2. С помощью программы blastn найти в нуклеотидном виде гомологи этой последовательности https://blast.ncbi.nlm.nih.gov/Blast.cgi
- 39. Задача 2 3. Открыть новый файл в программе UltraEdit
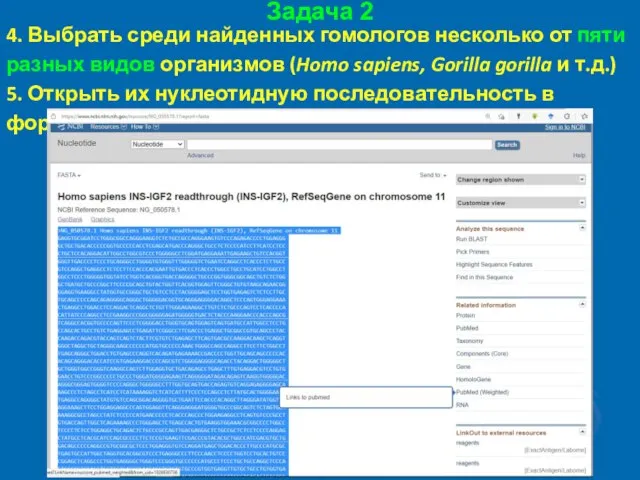
- 40. Задача 2 4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов (Homo sapiens, Gorilla
- 41. Задача 2 4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов (Homo sapiens, Gorilla
- 42. Задача 2 4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов (Homo sapiens, Gorilla
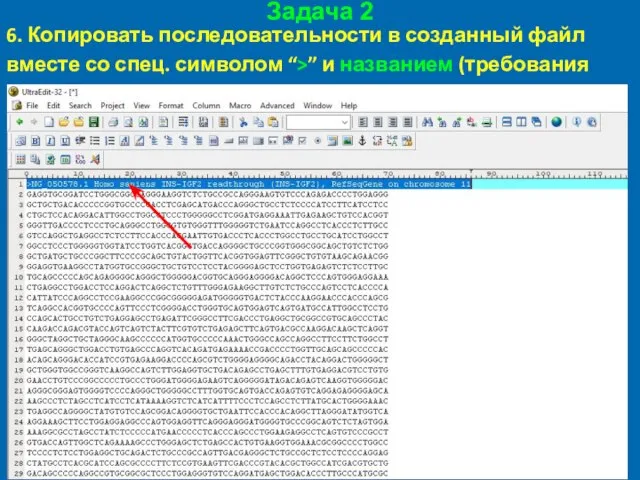
- 43. Задача 2 6. Копировать последовательности в созданный файл вместе со спец. символом “>” и названием (требования
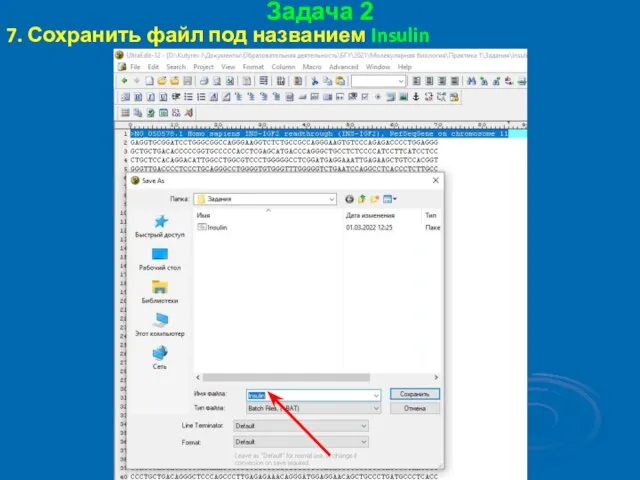
- 44. Задача 2 7. Сохранить файл под названием Insulin
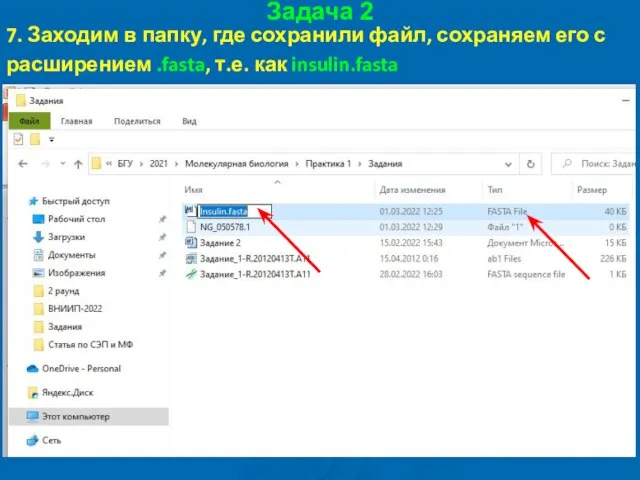
- 45. Задача 2 7. Заходим в папку, где сохранили файл, сохраняем его с расширением .fasta, т.е. как
- 46. Задача 2 8. Открыть файл с помощью программы MEGA5
- 47. Задача 2 8. Выровнять последовательности в программе MEGA5
- 48. Задача 2 8. Выровнять последовательности в программе MEGA5
- 49. Задача 2 8. Выровнять последовательности в программе MEGA5
- 50. Задача 2 9. Сохранить выравнивание в файл с названием insulin_alignment в формате FASTA
- 51. Задача 2 9. Сохранить выравнивание в файл с названием insulin_alignment в формате FASTA
- 52. Задача 2 9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
- 53. Задача 2 9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
- 54. Задача 2 9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
- 55. Задача 2 10. Смотрим расшифровку наилучшей модели внизу таблицы, необходимую для построения филогенетического дерева:
- 56. Задача 2 11. Сохраняем таблицу в формате Ecxel с именем insulin_best model
- 57. Задача 2 12. Построить филогенетическое дерево для выбранных нуклеотидных последовательностей
- 58. Задача 2 12. Указываем нужную (наилучшую) модель
- 59. Задача 2 12. При необходимости – другие параметры, указанные в наилучшей модели
- 60. Задача 2 12. Строим филогенетическое дерево для выбранных нуклеотидных последовательностей
- 61. Задача 2 12. Строим филогенетическое дерево для выбранных нуклеотидных последовательностей
- 62. Задача 2 13. Сохраняем текущую сессию под названием insulin_tree
- 64. Скачать презентацию

Слайд 3Выравнивание генетических последовательностей
В эволюции генетических последовательностей происходят как замены, так и вставки
Выравнивание генетических последовательностей
В эволюции генетических последовательностей происходят как замены, так и вставки

Слайд 4Выравнивание генетических последовательностей
Выравнивание генетических последовательностей

Слайд 5Выравнивание генетических последовательностей
Выравнивание генетических последовательностей

Слайд 6Выравнивание генетических последовательностей
Clustal -- это одна из самых широко используемых компьютерных программ
Выравнивание генетических последовательностей
Clustal -- это одна из самых широко используемых компьютерных программ

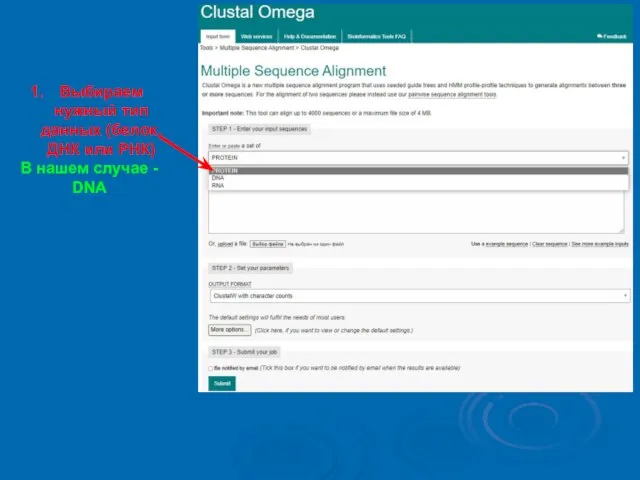
Слайд 7Выбираем нужный тип данных (белок, ДНК или РНК)
В нашем случае - DNA
Выбираем нужный тип данных (белок, ДНК или РНК)
В нашем случае - DNA
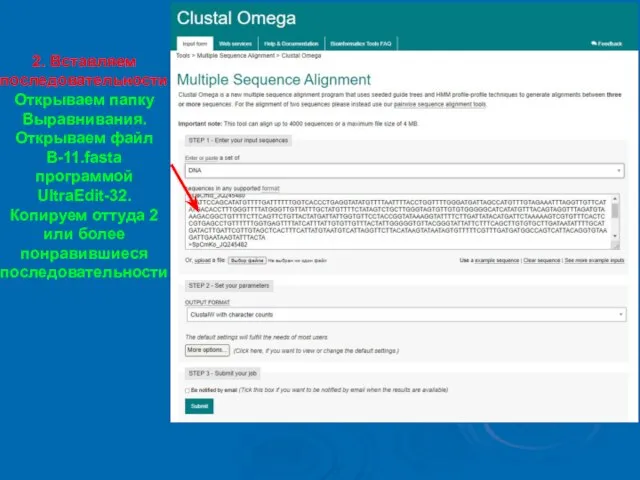
Слайд 82. Вставляем последовательности
Открываем папку Выравнивания. Открываем файл В-11.fasta программой UltraEdit-32.
Копируем оттуда 2
2. Вставляем последовательности
Открываем папку Выравнивания. Открываем файл В-11.fasta программой UltraEdit-32.
Копируем оттуда 2
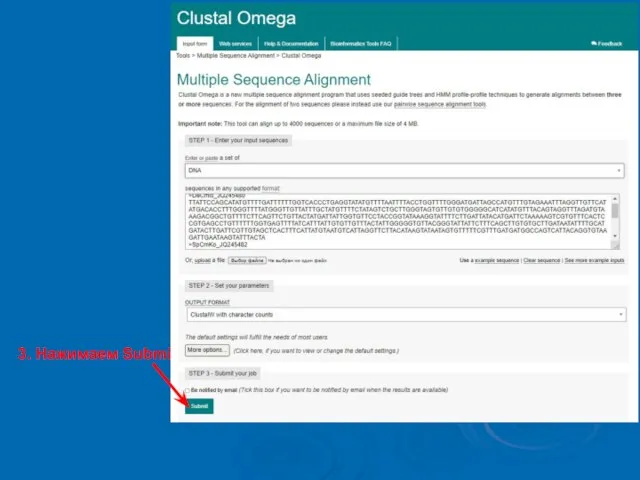
Слайд 93. Нажимаем Submit
3. Нажимаем Submit
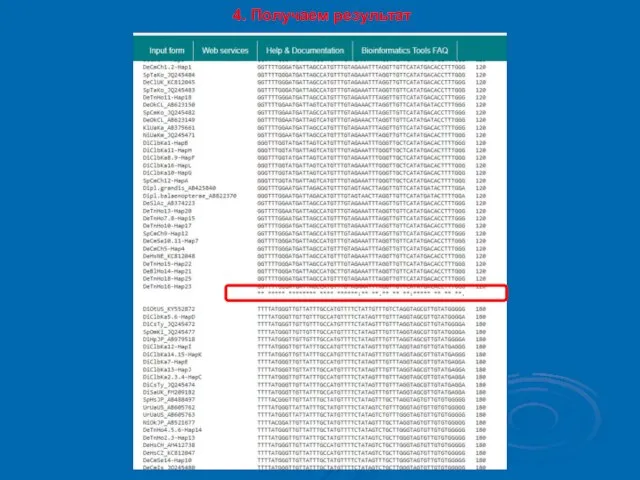
Слайд 104. Получаем результат
4. Получаем результат
Слайд 115. Интерпретация
Звездочка (*) – различия по данной позиции (нуклеотид или аминокислота) отсутствуют
5. Интерпретация
Звездочка (*) – различия по данной позиции (нуклеотид или аминокислота) отсутствуют

Слайд 126. Здесь же можно посмотреть предварительное филогенетическое дерево
6. Здесь же можно посмотреть предварительное филогенетическое дерево

Слайд 14BLAST
BLAST (англ. Basic Local Alignment Search Tool — средство поиска основного локального
BLAST
BLAST (англ. Basic Local Alignment Search Tool — средство поиска основного локального

Слайд 15Классификация программ серии BLAST
Нуклеотидные
предназначены для сравнения изучаемой нуклеотидной последовательности с базой данных
Классификация программ серии BLAST
Нуклеотидные
предназначены для сравнения изучаемой нуклеотидной последовательности с базой данных

Слайд 16Классификация программ серии BLAST
Белковые
предназначены для сравнения изучаемой аминокислотной последовательности белка с имеющейся
Классификация программ серии BLAST
Белковые
предназначены для сравнения изучаемой аминокислотной последовательности белка с имеющейся

Слайд 17Классификация программ серии BLAST
Транслирующие
способны транслировать нуклеотидные последовательности в аминокислотные:
blastx — переводит изучаемую нуклеотидную
Классификация программ серии BLAST
Транслирующие
способны транслировать нуклеотидные последовательности в аминокислотные:
blastx — переводит изучаемую нуклеотидную

Слайд 18Классификация программ серии BLAST
Геномные
предназначены для сравнения изучаемой нуклеотидной последовательности с базой данных
Классификация программ серии BLAST
Геномные
предназначены для сравнения изучаемой нуклеотидной последовательности с базой данных

Слайд 19Переходим в сервис BLAST Национального центра биотехнологической информации США (NCBI) по ссылке:
https://blast.ncbi.nlm.nih.gov/Blast.cgi
Переходим в сервис BLAST Национального центра биотехнологической информации США (NCBI) по ссылке:
https://blast.ncbi.nlm.nih.gov/Blast.cgi
Слайд 20blastn
tblastn
blastx
blastp
blastn
tblastn
blastx
blastp
Слайд 21Задача 1. Форма отчета
Каждый лично на своем компьютере делает скриншот/фото (так, чтобы
Задача 1. Форма отчета
Каждый лично на своем компьютере делает скриншот/фото (так, чтобы

Слайд 22Задача 1
Открыть с помощью программы Chromas файл с хроматограммой Задание_1-R.20120413T.A11 из
Задача 1
Открыть с помощью программы Chromas файл с хроматограммой Задание_1-R.20120413T.A11 из

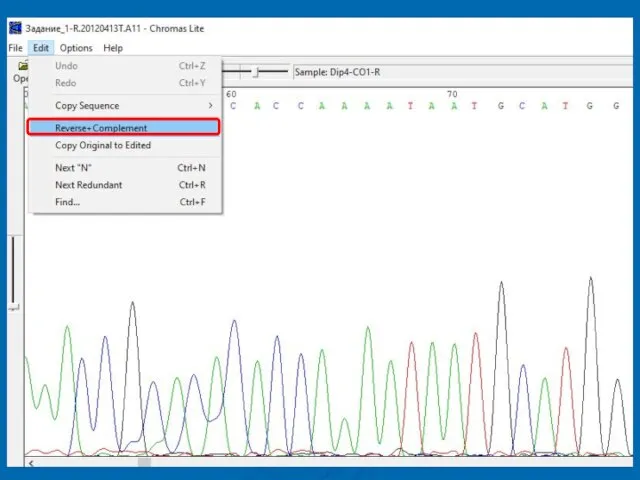
Слайд 24Задача 1
4. Вместо символов N поставить соответствующие хроматограмме нуклеотиды (A, T, G
Задача 1
4. Вместо символов N поставить соответствующие хроматограмме нуклеотиды (A, T, G
Слайд 25Задача 1
4. Вместо символов N поставить соответствующие хроматограмме нуклеотиды (A, T, G
Задача 1
4. Вместо символов N поставить соответствующие хроматограмме нуклеотиды (A, T, G
Слайд 26Задача 1
5. Сохранить отредактированную последовательность в новый файл в формате FASTA:
Задача 1
5. Сохранить отредактированную последовательность в новый файл в формате FASTA:
Слайд 27Задача 1
6. Открыть сохраненный файл в программе UltraEdit-32
7. Копировать последовательность нуклеотидов:
Задача 1
6. Открыть сохраненный файл в программе UltraEdit-32
7. Копировать последовательность нуклеотидов:
Слайд 28Задача 1
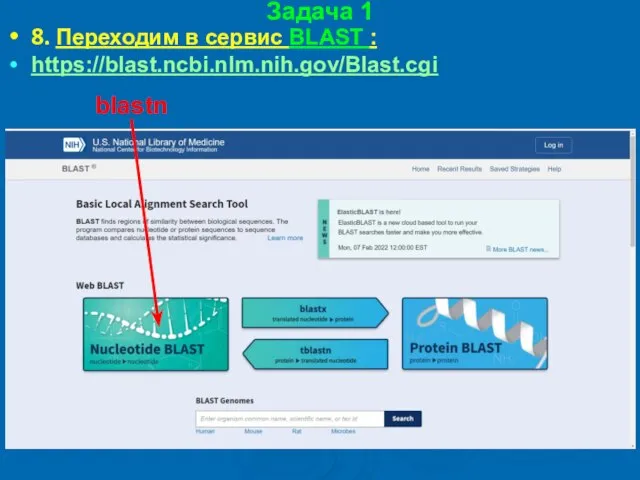
8. Переходим в сервис BLAST :
https://blast.ncbi.nlm.nih.gov/Blast.cgi
blastn
Задача 1
8. Переходим в сервис BLAST :
https://blast.ncbi.nlm.nih.gov/Blast.cgi
blastn
Слайд 29Задача 1
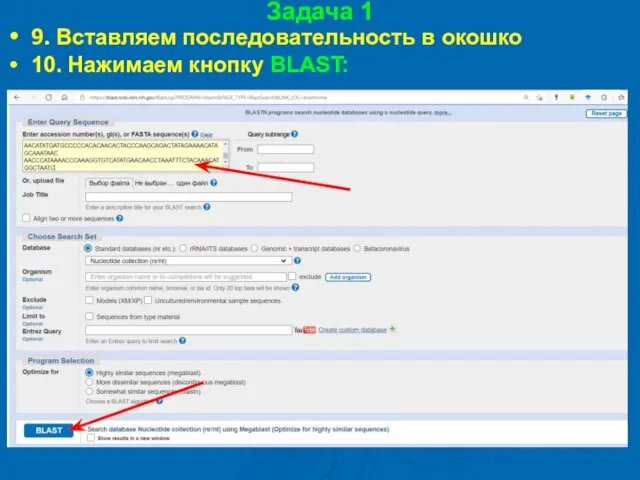
9. Вставляем последовательность в окошко
10. Нажимаем кнопку BLAST:
Задача 1
9. Вставляем последовательность в окошко
10. Нажимаем кнопку BLAST:
Слайд 30Задача 1
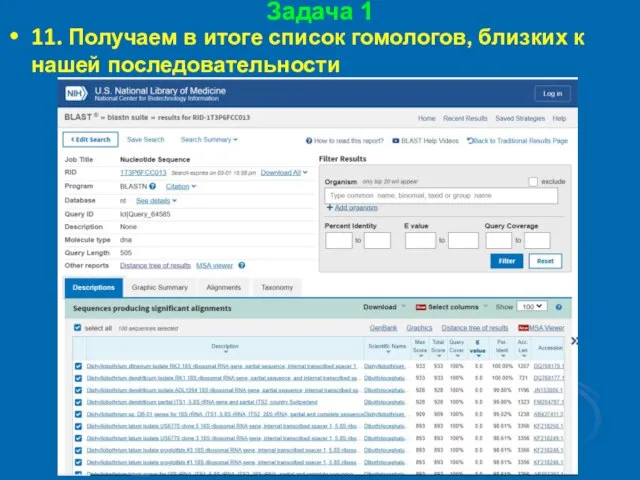
11. Получаем в итоге список гомологов, близких к нашей последовательности
Задача 1
11. Получаем в итоге список гомологов, близких к нашей последовательности
Слайд 31Задача 1. Форма отчета
Каждый лично на своем компьютере делает скриншот/фото (так, чтобы
Задача 1. Форма отчета
Каждый лично на своем компьютере делает скриншот/фото (так, чтобы

Слайд 32Задача 2. Форма отчета
Каждый лично отправляет мне на почту [email protected] :
1) файл
Задача 2. Форма отчета
Каждый лично отправляет мне на почту [email protected] :
1) файл

Слайд 33Задача 2
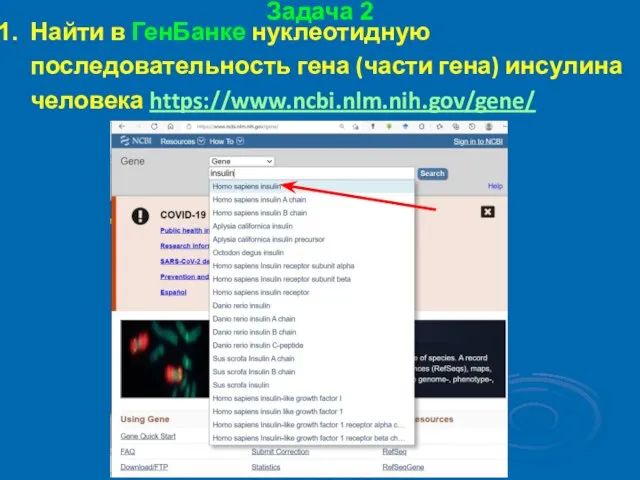
Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
Задача 2
Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
Слайд 34Задача 2
Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
Задача 2
Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
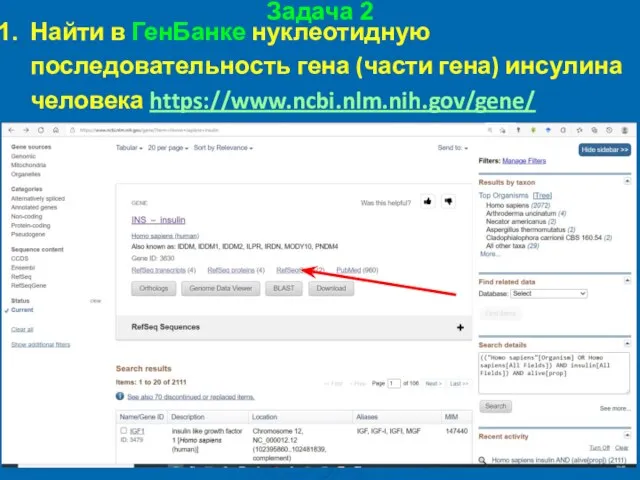
Слайд 35Задача 2
Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
Задача 2
Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
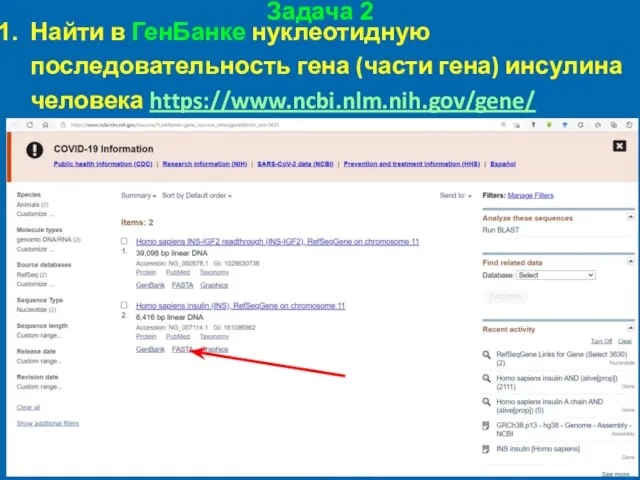
Слайд 36Задача 2
Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
Задача 2
Найти в ГенБанке нуклеотидную последовательность гена (части гена) инсулина человека https://www.ncbi.nlm.nih.gov/gene/
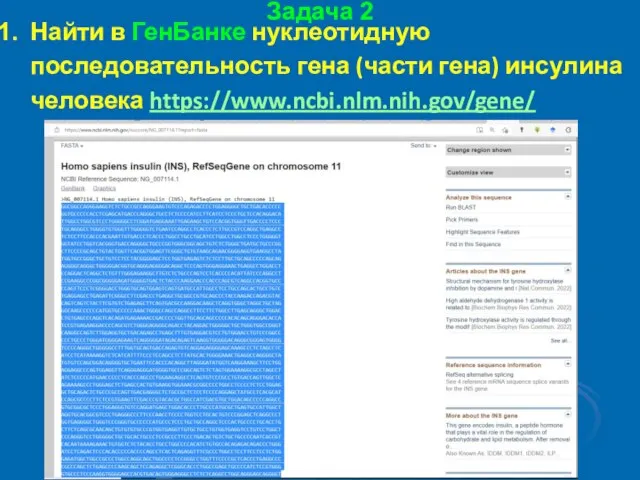
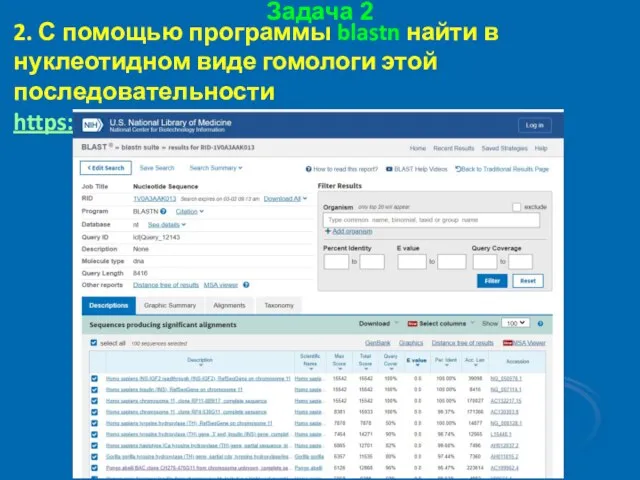
Слайд 37Задача 2
2. С помощью программы blastn найти в нуклеотидном виде гомологи этой
Задача 2
2. С помощью программы blastn найти в нуклеотидном виде гомологи этой
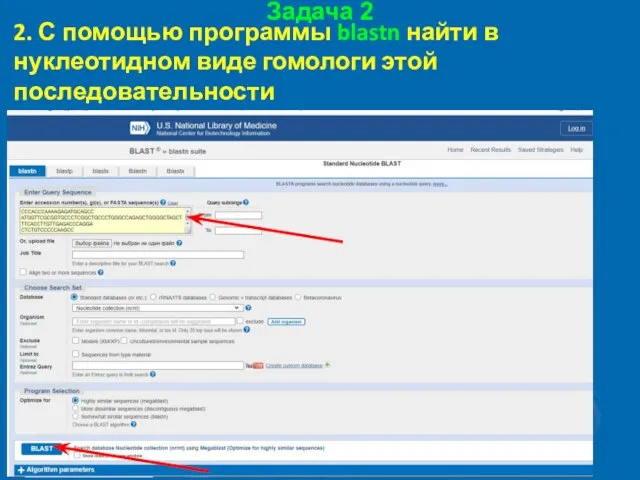
Слайд 38Задача 2
2. С помощью программы blastn найти в нуклеотидном виде гомологи этой
Задача 2
2. С помощью программы blastn найти в нуклеотидном виде гомологи этой
Слайд 39Задача 2
3. Открыть новый файл в программе UltraEdit
Задача 2
3. Открыть новый файл в программе UltraEdit
Слайд 40Задача 2
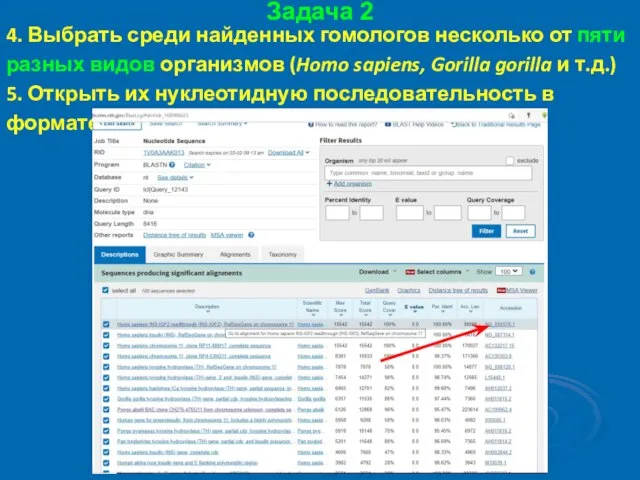
4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов
Задача 2
4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов
Слайд 41Задача 2
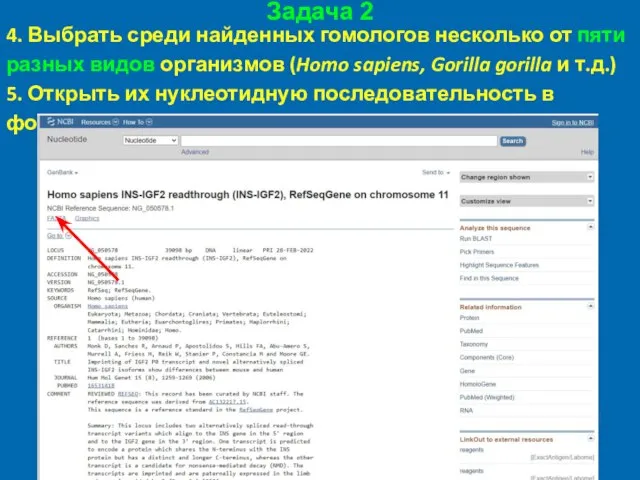
4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов
Задача 2
4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов
Слайд 42Задача 2
4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов
Задача 2
4. Выбрать среди найденных гомологов несколько от пяти разных видов организмов
Слайд 43Задача 2
6. Копировать последовательности в созданный файл вместе со спец. символом “>”
Задача 2
6. Копировать последовательности в созданный файл вместе со спец. символом “>”
Слайд 44Задача 2
7. Сохранить файл под названием Insulin
Задача 2
7. Сохранить файл под названием Insulin
Слайд 45Задача 2
7. Заходим в папку, где сохранили файл, сохраняем его с расширением
Задача 2
7. Заходим в папку, где сохранили файл, сохраняем его с расширением
Слайд 46Задача 2
8. Открыть файл с помощью программы MEGA5
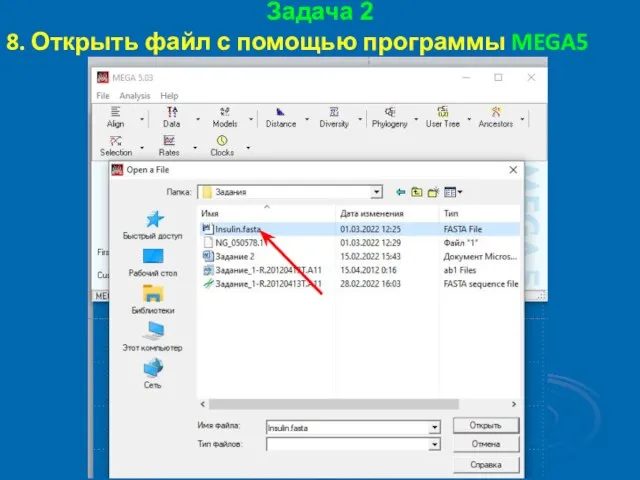
Задача 2
8. Открыть файл с помощью программы MEGA5
Слайд 47Задача 2
8. Выровнять последовательности в программе MEGA5
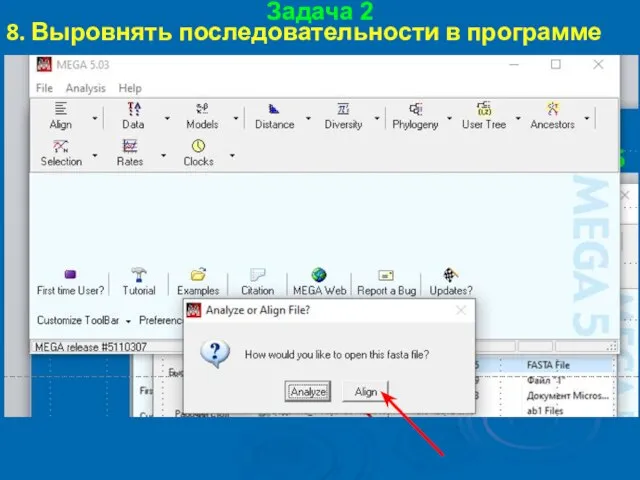
Задача 2
8. Выровнять последовательности в программе MEGA5
Слайд 48Задача 2
8. Выровнять последовательности в программе MEGA5
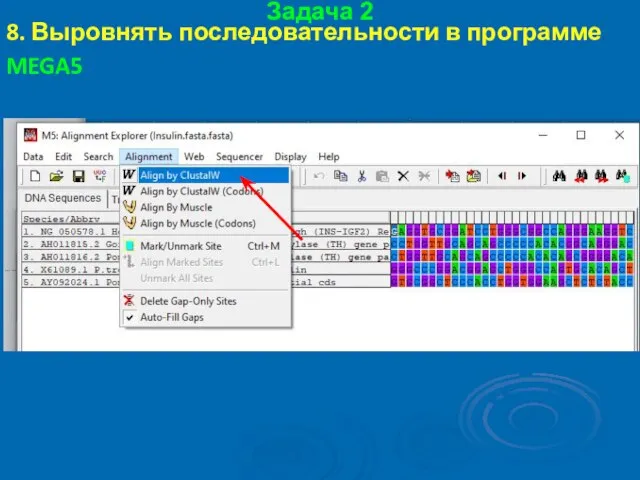
Задача 2
8. Выровнять последовательности в программе MEGA5
Слайд 49Задача 2
8. Выровнять последовательности в программе MEGA5
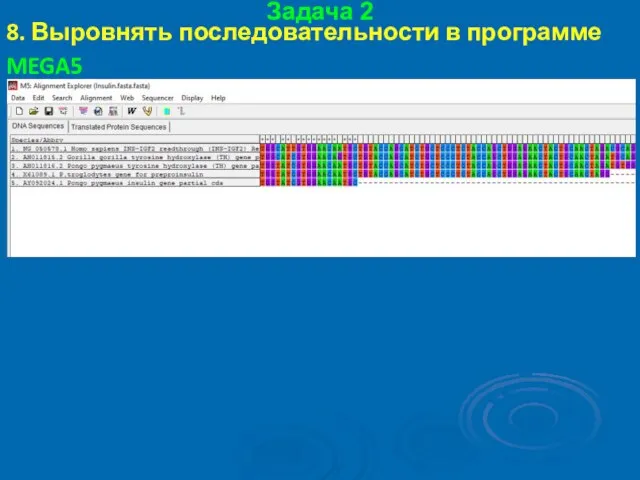
Задача 2
8. Выровнять последовательности в программе MEGA5
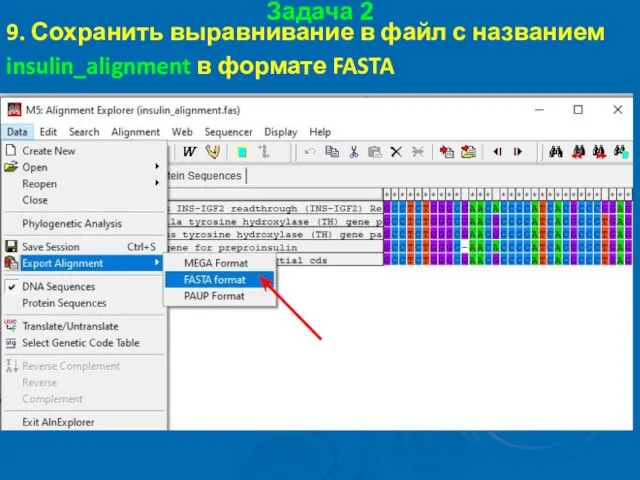
Слайд 50Задача 2
9. Сохранить выравнивание в файл с названием insulin_alignment в формате FASTA
Задача 2
9. Сохранить выравнивание в файл с названием insulin_alignment в формате FASTA
Слайд 51Задача 2
9. Сохранить выравнивание в файл с названием insulin_alignment в формате FASTA
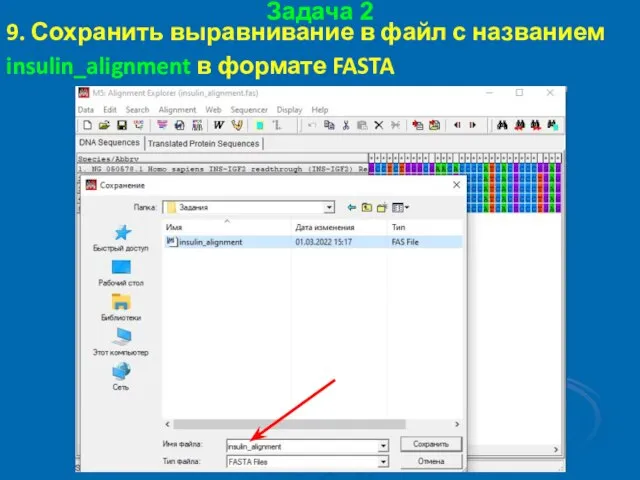
Задача 2
9. Сохранить выравнивание в файл с названием insulin_alignment в формате FASTA
Слайд 52Задача 2
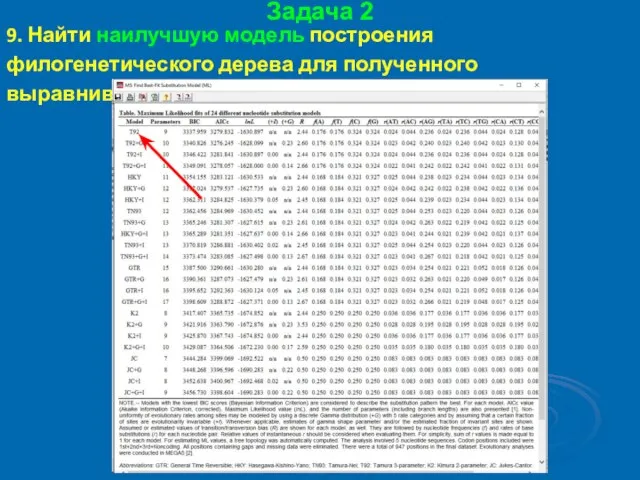
9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
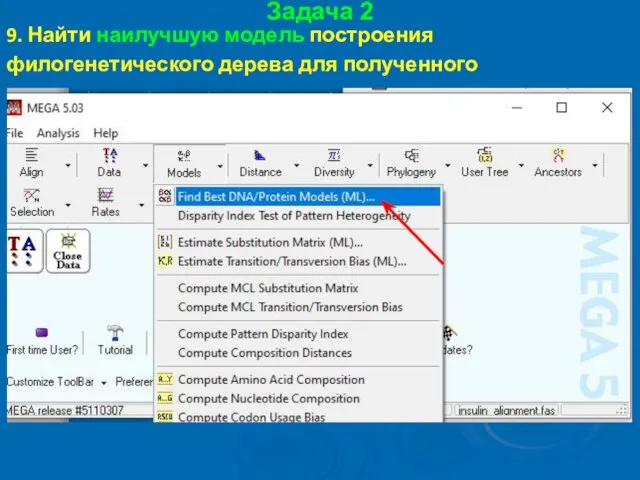
Задача 2
9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
Слайд 53Задача 2
9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
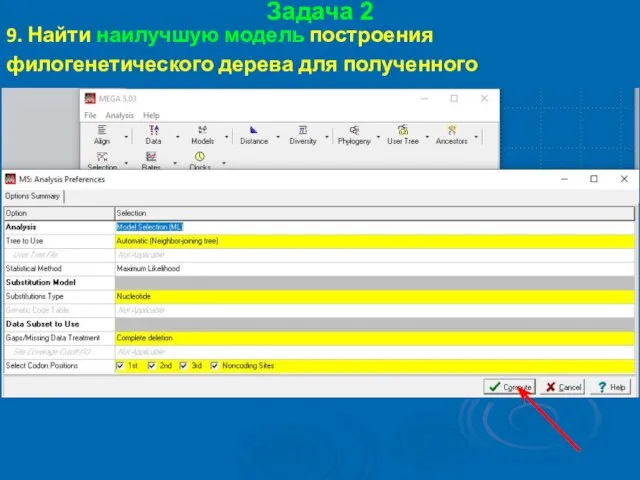
Задача 2
9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
Слайд 54Задача 2
9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
Задача 2
9. Найти наилучшую модель построения филогенетического дерева для полученного выравнивания
Слайд 55Задача 2
10. Смотрим расшифровку наилучшей модели внизу таблицы, необходимую для построения филогенетического
Задача 2
10. Смотрим расшифровку наилучшей модели внизу таблицы, необходимую для построения филогенетического

Слайд 56Задача 2
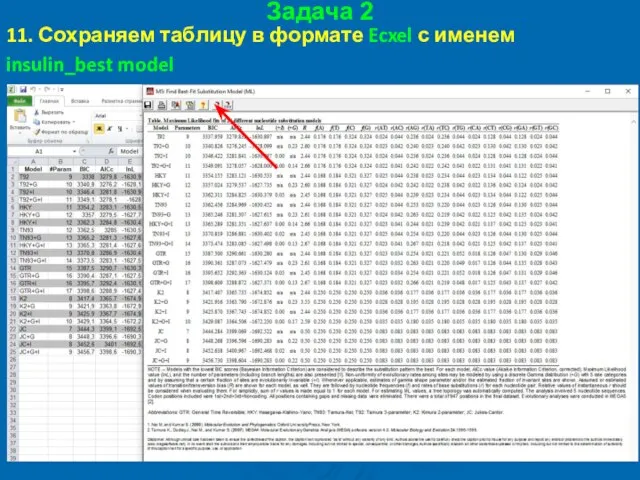
11. Сохраняем таблицу в формате Ecxel с именем
insulin_best model
Задача 2
11. Сохраняем таблицу в формате Ecxel с именем
insulin_best model
Слайд 57Задача 2
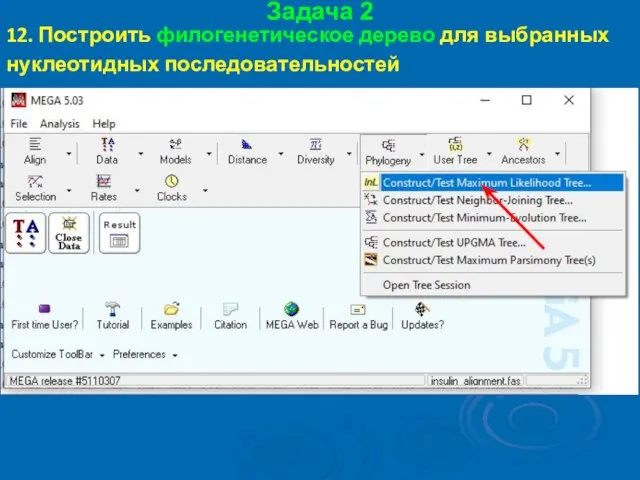
12. Построить филогенетическое дерево для выбранных нуклеотидных последовательностей
Задача 2
12. Построить филогенетическое дерево для выбранных нуклеотидных последовательностей
Слайд 58Задача 2
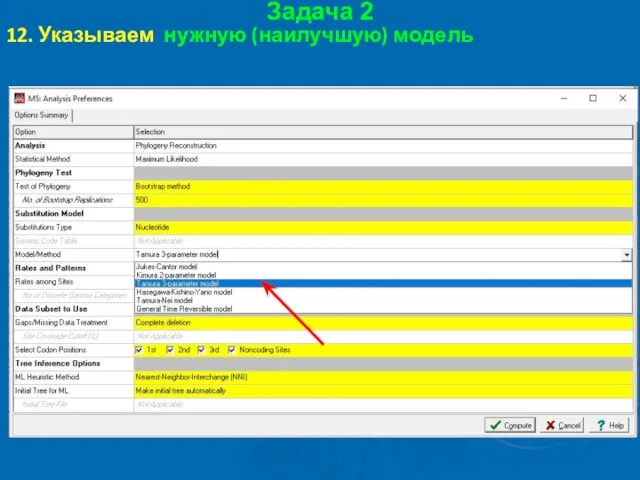
12. Указываем нужную (наилучшую) модель
Задача 2
12. Указываем нужную (наилучшую) модель
Слайд 59Задача 2
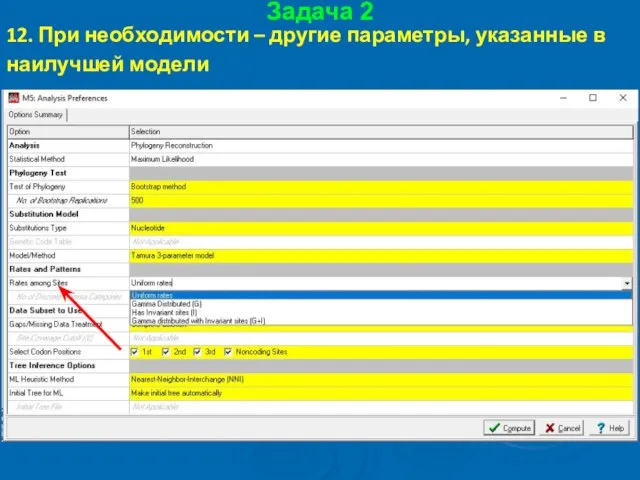
12. При необходимости – другие параметры, указанные в наилучшей модели
Задача 2
12. При необходимости – другие параметры, указанные в наилучшей модели
Слайд 60Задача 2
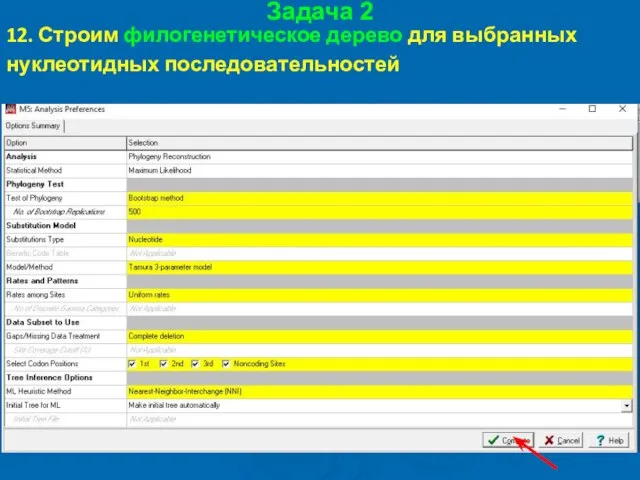
12. Строим филогенетическое дерево для выбранных нуклеотидных последовательностей
Задача 2
12. Строим филогенетическое дерево для выбранных нуклеотидных последовательностей
Слайд 61Задача 2
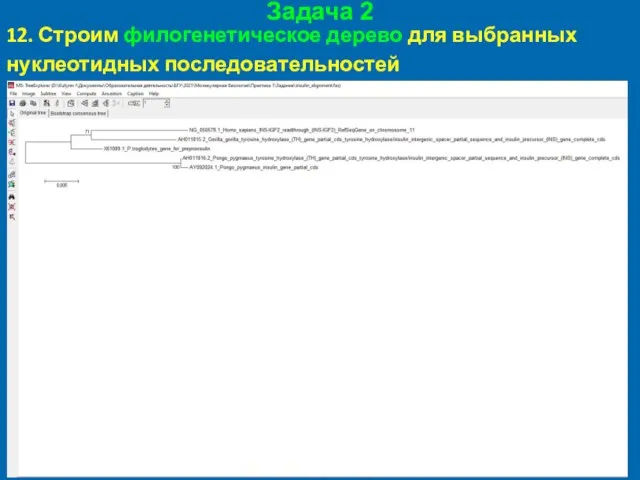
12. Строим филогенетическое дерево для выбранных нуклеотидных последовательностей
Задача 2
12. Строим филогенетическое дерево для выбранных нуклеотидных последовательностей
Слайд 62Задача 2
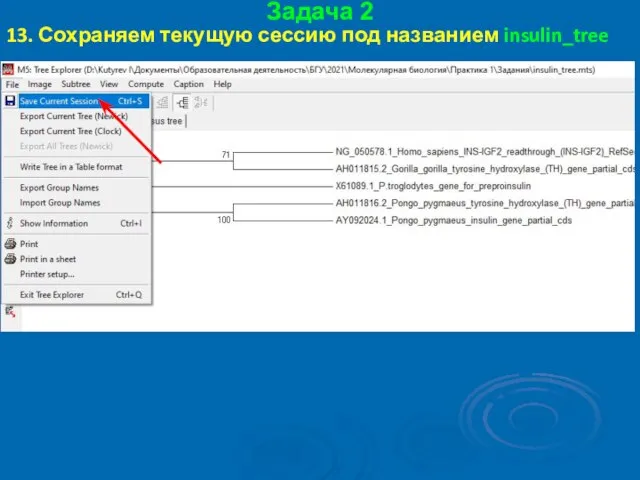
13. Сохраняем текущую сессию под названием insulin_tree
Задача 2
13. Сохраняем текущую сессию под названием insulin_tree
 Современное представление о механизмах памяти
Современное представление о механизмах памяти Презентация на тему РОГАТЫЕ НАСЕКОМЫЕ
Презентация на тему РОГАТЫЕ НАСЕКОМЫЕ  6-Б 12.10 (1)
6-Б 12.10 (1) Дарвин и теория эволюции
Дарвин и теория эволюции Как возникла жизнь: биологическая эволюция или креационизм. Лекция 7
Как возникла жизнь: биологическая эволюция или креационизм. Лекция 7 Водоросли
Водоросли Эко косметика
Эко косметика Мышцы тазового дна
Мышцы тазового дна Класс Млекопитающие. Отряды Грызуны, Зайцеобразные. 7 класс
Класс Млекопитающие. Отряды Грызуны, Зайцеобразные. 7 класс Биологические аспекты адаптации населения к условиям жизнедеятельности
Биологические аспекты адаптации населения к условиям жизнедеятельности ZhIRORASTVORIMYE_VITAMINY-15
ZhIRORASTVORIMYE_VITAMINY-15 Ядро эукариотической клетки
Ядро эукариотической клетки Водоросли. Строение клетки. Размножение
Водоросли. Строение клетки. Размножение Ткани растений. 10 класс
Ткани растений. 10 класс Удивительные растения планеты
Удивительные растения планеты Венерина мухоловка
Венерина мухоловка Биология зависимости. Химическая зависимость. Нейропсихиатрия и биология зависимости. Занятие 5
Биология зависимости. Химическая зависимость. Нейропсихиатрия и биология зависимости. Занятие 5 Сердечно-сосудистая система у домашних млекопитающих животных
Сердечно-сосудистая система у домашних млекопитающих животных Собираем грибы
Собираем грибы Биология клетки. Общие черты строения типичной клетки. Особенности строения клеток в связи с их функцией
Биология клетки. Общие черты строения типичной клетки. Особенности строения клеток в связи с их функцией Отдел Сложноцветные
Отдел Сложноцветные Тайга, животный мир
Тайга, животный мир Презентация на тему Состав почвы
Презентация на тему Состав почвы  эволюция пищеварения животных
эволюция пищеварения животных Мероприятия по охране атмосферы от загрязнения. Источники загрязнения воздуха и их последствия. Растения – очистители воздуха
Мероприятия по охране атмосферы от загрязнения. Источники загрязнения воздуха и их последствия. Растения – очистители воздуха § 37. Строение и деятельность внутренних органов 7 класс биология
§ 37. Строение и деятельность внутренних органов 7 класс биология Лиственница
Лиственница Формы естественного отбора
Формы естественного отбора