- Обработка протеомных данных (массспектрометрия)

Содержание
- 2. Подходы к анализу в протеомной масс-спектрометрии I. “Восходящий” анализ – Bottom-up гидролиз белка/белков, масс-спектрометрия малых пептидных
- 3. Два главных подхода к протеомному анализу
- 4. Top-down: изучение интактных белков 1). Позволяет изучать индивидуальные протеины. 2). Является низкопроизводительным подходом. Исключение: изучение белкового
- 5. Bottom-up Данный подход преобладает в протеомных исследованиях. Существует в двух вариантах: 1. Выделение одного/нескольких белков (PAGE),
- 6. Типы протеомного исследования 1. Определение наличия в образце белков с известной последовательностью (“ресеквенирование” белков). 2. Определение
- 7. Определение белков de novo Применяется на основе bottom-up подхода (анализ пептидных спектров). В данном случае требуется
- 9. Основа анализа масс-спектров – определение типов образующихся ионов В подавляющем большинстве методов МС в результате ионизации
- 10. Главные типы ионов, образующихся в протеомной МС Существует три вида связей в пептидах, по которым происходит
- 11. Дополнительные пептидные ионы При более жёсткой ионизации наряду с главными ионами происходит образование саттелитных ионов (d-,
- 12. Главные и саттелитные пептидные ионы
- 13. Связь между массой главного иона и массами аминокислотных остатков иона Масса образующихся главных ионов определяется суммой
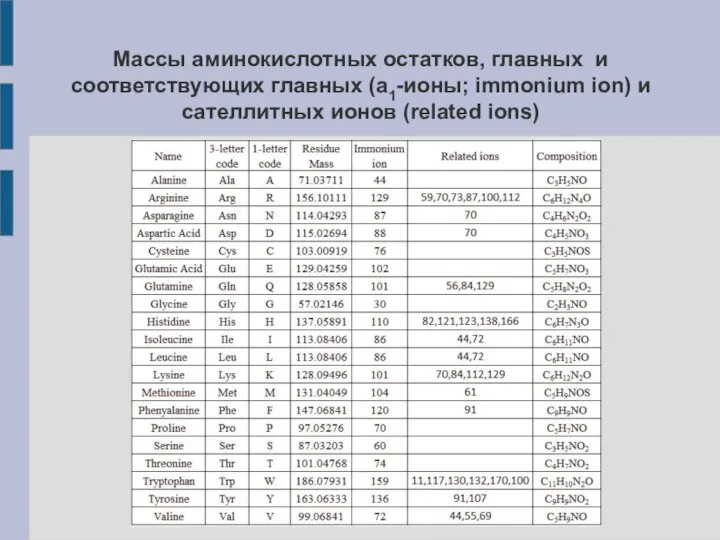
- 14. Массы аминокислотных остатков, главных и соответствующих главных (a1-ионы; immonium ion) и сателлитных ионов (related ions)
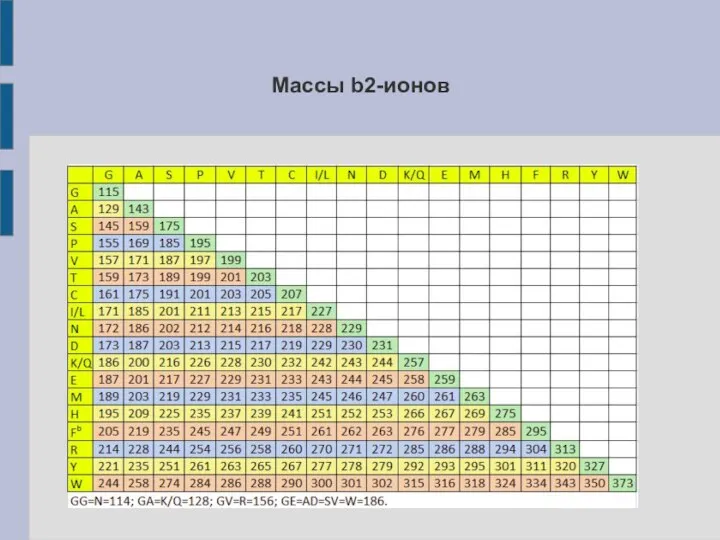
- 15. Массы b2-ионов
- 16. “Правила” ионизации пептидов 1). Подавляющее большинство ионов приходится на b-, y- и a-ионы. а-Ионы образуются из
- 17. Количественный протеомный анализ Позволяет оценивать относительные количества одинаковых белков их разных источников в результате анализа смешанного
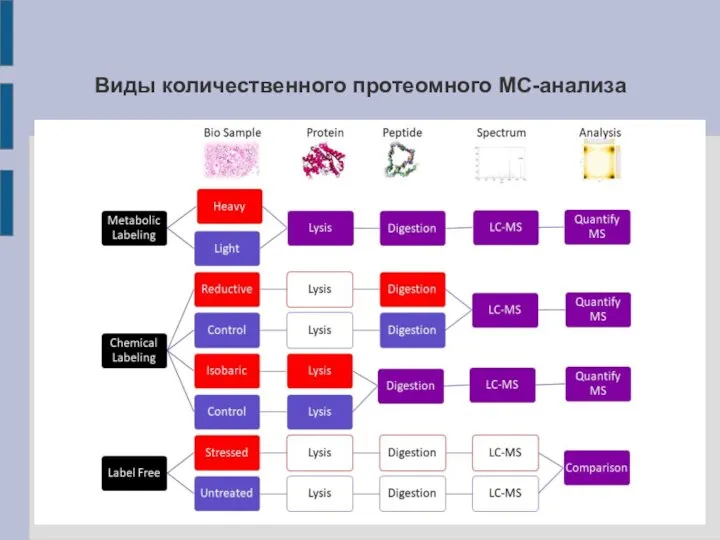
- 18. Виды количественного протеомного МС-анализа
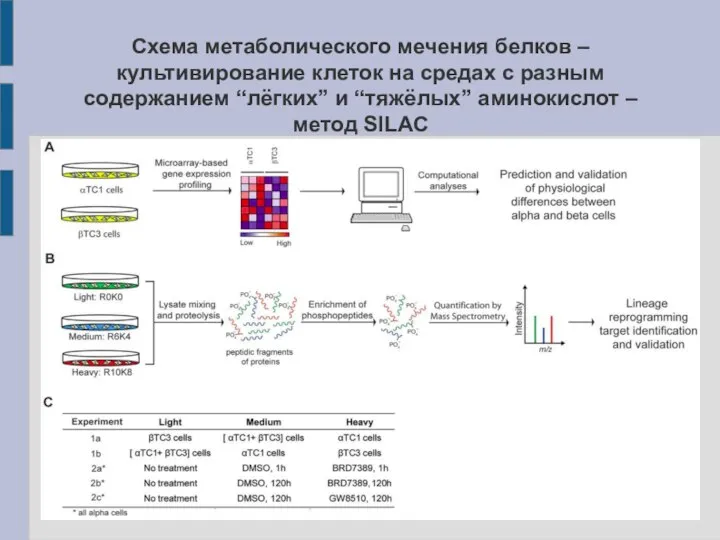
- 19. Схема метаболического мечения белков – культивирование клеток на средах с разным содержанием “лёгких” и “тяжёлых” аминокислот
- 20. Форматы представления МС-данных 1. “Родные” форматы приборов TDF (Bruker), t2d (ABI), lcd (Shimadzu), tdc (Physical Electronics)
- 21. Примеры представления данных из mzXML файлов в разных программах (a,b – InsilicosViewer; c – mzXML Spectrum
- 22. Анализ результатов МС-эксперимента 1. Поиск по базам данных масс-спектров пептидных фрагментов записей, отвечающих (с учётом посттрансляционных
- 23. Пример результатов анализа масс-спектра с помощью инструмента MS-Fit онлайн-ресурса ProteinProspector
- 24. FASTA – универсальный формат представления первичной структуры белков и нуклеиновых кислот >BBB04705.1 M1 protein [Influenza A
- 25. Обозначение аминокислот в формате FASTA
- 26. Трёхмерная структура гемагглютинина H1, полученная с помощью средств базы данных PDB (разным цветом обозначены разные участки
- 28. Скачать презентацию
Слайд 2Подходы к анализу в протеомной масс-спектрометрии
I. “Восходящий” анализ – Bottom-up
гидролиз белка/белков, масс-спектрометрия
Подходы к анализу в протеомной масс-спектрометрии
I. “Восходящий” анализ – Bottom-up
гидролиз белка/белков, масс-спектрометрия

Слайд 3Два главных подхода к протеомному анализу
Два главных подхода к протеомному анализу
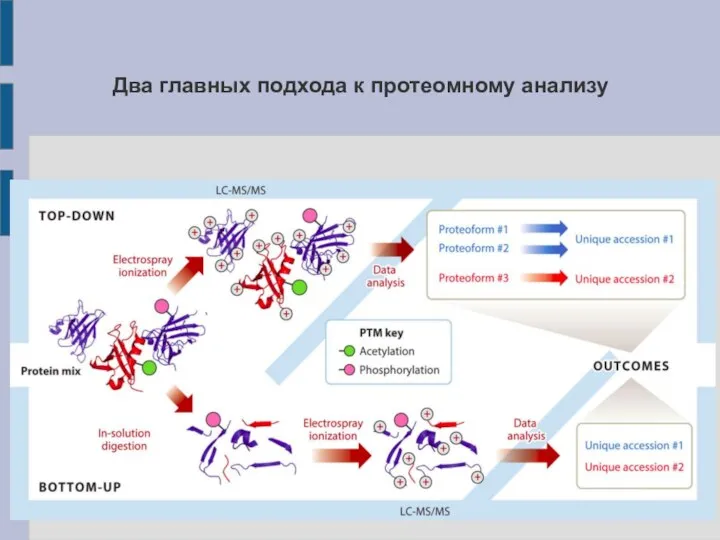
Слайд 4Top-down: изучение интактных белков
1). Позволяет изучать индивидуальные протеины.
2). Является низкопроизводительным подходом.
Исключение: изучение
Top-down: изучение интактных белков
1). Позволяет изучать индивидуальные протеины.
2). Является низкопроизводительным подходом.
Исключение: изучение

Слайд 5Bottom-up
Данный подход преобладает в протеомных исследованиях.
Существует в двух вариантах:
1. Выделение одного/нескольких белков
Bottom-up
Данный подход преобладает в протеомных исследованиях.
Существует в двух вариантах:
1. Выделение одного/нескольких белков
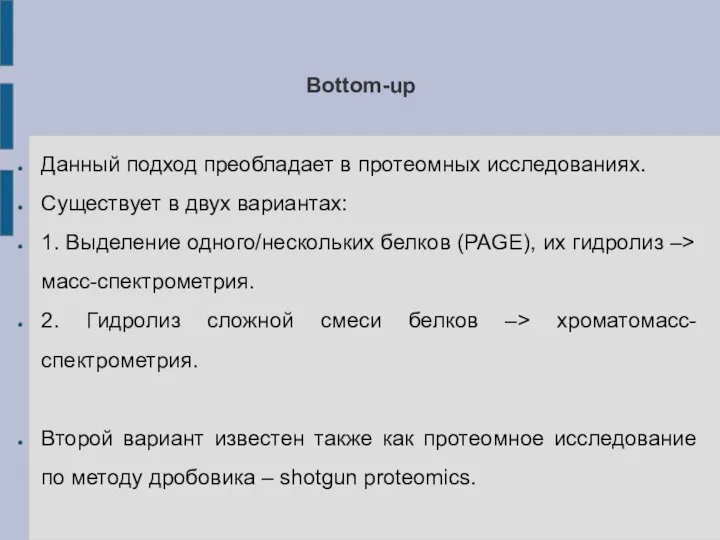
Слайд 6Типы протеомного исследования
1. Определение наличия в образце белков с известной последовательностью (“ресеквенирование”
Типы протеомного исследования
1. Определение наличия в образце белков с известной последовательностью (“ресеквенирование”

Слайд 7Определение белков de novo
Применяется на основе bottom-up подхода (анализ пептидных спектров). В
Определение белков de novo
Применяется на основе bottom-up подхода (анализ пептидных спектров). В

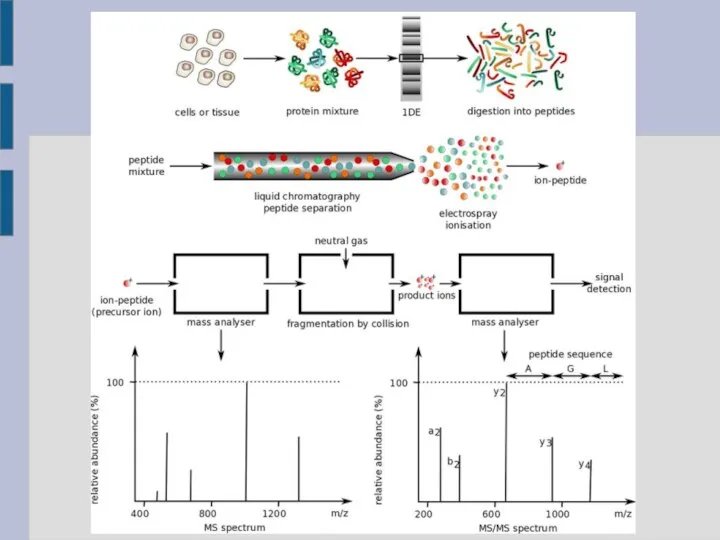
Слайд 9Основа анализа масс-спектров – определение типов образующихся ионов
В подавляющем большинстве методов МС
Основа анализа масс-спектров – определение типов образующихся ионов
В подавляющем большинстве методов МС

Слайд 10Главные типы ионов, образующихся в протеомной МС
Существует три вида связей в пептидах,
Главные типы ионов, образующихся в протеомной МС
Существует три вида связей в пептидах,

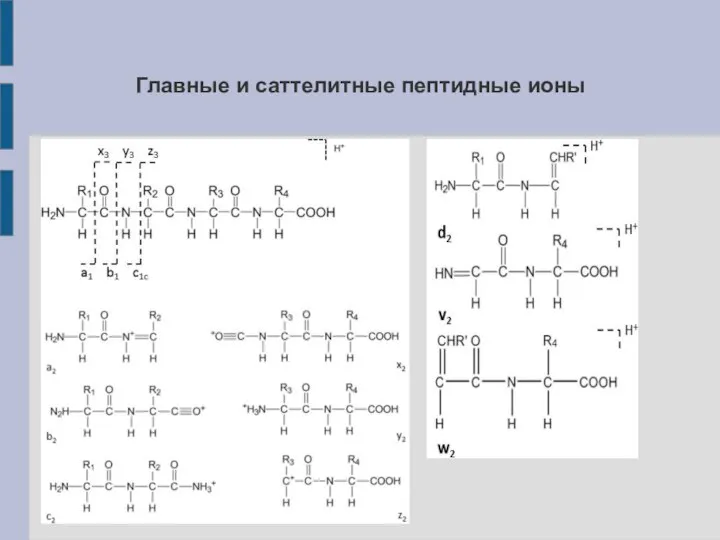
Слайд 11Дополнительные пептидные ионы
При более жёсткой ионизации наряду с главными ионами происходит образование
Дополнительные пептидные ионы
При более жёсткой ионизации наряду с главными ионами происходит образование

Слайд 12Главные и саттелитные пептидные ионы
Главные и саттелитные пептидные ионы
Слайд 13Связь между массой главного иона и массами аминокислотных остатков иона
Масса образующихся главных
Связь между массой главного иона и массами аминокислотных остатков иона
Масса образующихся главных

Слайд 14Массы аминокислотных остатков, главных и соответствующих главных (a1-ионы; immonium ion) и сателлитных
Массы аминокислотных остатков, главных и соответствующих главных (a1-ионы; immonium ion) и сателлитных
Слайд 15Массы b2-ионов
Массы b2-ионов
Слайд 16“Правила” ионизации пептидов
1). Подавляющее большинство ионов приходится на b-, y- и
“Правила” ионизации пептидов
1). Подавляющее большинство ионов приходится на b-, y- и

Слайд 17Количественный протеомный анализ
Позволяет оценивать относительные количества одинаковых белков их разных источников в
Количественный протеомный анализ
Позволяет оценивать относительные количества одинаковых белков их разных источников в

Слайд 18Виды количественного протеомного МС-анализа
Виды количественного протеомного МС-анализа
Слайд 19Схема метаболического мечения белков – культивирование клеток на средах с разным содержанием
Схема метаболического мечения белков – культивирование клеток на средах с разным содержанием
Слайд 20Форматы представления МС-данных
1. “Родные” форматы приборов
TDF (Bruker), t2d (ABI), lcd (Shimadzu), tdc
Форматы представления МС-данных
1. “Родные” форматы приборов
TDF (Bruker), t2d (ABI), lcd (Shimadzu), tdc

Слайд 21Примеры представления данных из mzXML файлов в разных программах (a,b – InsilicosViewer;
Примеры представления данных из mzXML файлов в разных программах (a,b – InsilicosViewer;
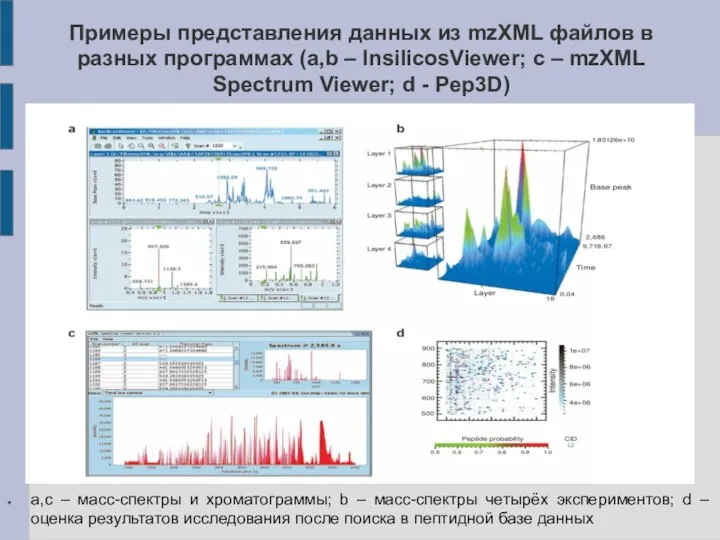
Слайд 22Анализ результатов МС-эксперимента
1. Поиск по базам данных масс-спектров пептидных фрагментов записей, отвечающих
Анализ результатов МС-эксперимента
1. Поиск по базам данных масс-спектров пептидных фрагментов записей, отвечающих

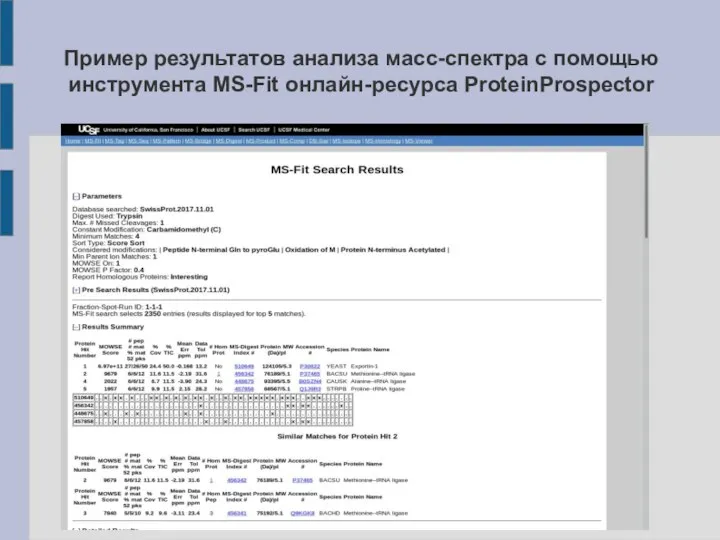
Слайд 23Пример результатов анализа масс-спектра с помощью инструмента MS-Fit онлайн-ресурса ProteinProspector
Пример результатов анализа масс-спектра с помощью инструмента MS-Fit онлайн-ресурса ProteinProspector
Слайд 24FASTA – универсальный формат представления первичной структуры белков и нуклеиновых кислот
>BBB04705.1 M1
FASTA – универсальный формат представления первичной структуры белков и нуклеиновых кислот
>BBB04705.1 M1

Слайд 25Обозначение аминокислот в формате FASTA
Обозначение аминокислот в формате FASTA
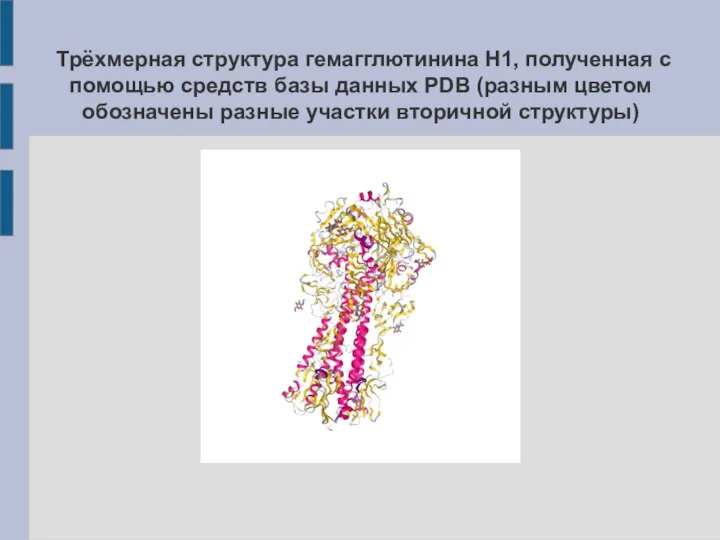
Слайд 26Трёхмерная структура гемагглютинина H1, полученная с помощью средств базы данных PDB (разным
Трёхмерная структура гемагглютинина H1, полученная с помощью средств базы данных PDB (разным
 Лекция 3
Лекция 3 Вегетативная нервная система
Вегетативная нервная система Перелётные птицы
Перелётные птицы Вегетативный способ размножения растений корнями и листьями
Вегетативный способ размножения растений корнями и листьями Эволюция человека и обезьян шла параллельно друг другу, а не последовательно
Эволюция человека и обезьян шла параллельно друг другу, а не последовательно Перепончатокрылые. Общая характеристика
Перепончатокрылые. Общая характеристика Витамин Вс
Витамин Вс Микологические исследования съедобных грибов на территории Сыктывдинского лесничества
Микологические исследования съедобных грибов на территории Сыктывдинского лесничества Всё о кошках и моей Муси…
Всё о кошках и моей Муси… Методы микроскопии
Методы микроскопии Вирусы – неклеточные формы жизни
Вирусы – неклеточные формы жизни Состав и биологическая активность фармацевтических препаратов, содержащих флавоноиды
Состав и биологическая активность фармацевтических препаратов, содержащих флавоноиды Своеобразие органического мира Австралии. Домашнее задание
Своеобразие органического мира Австралии. Домашнее задание Породы домашних животных
Породы домашних животных Строение и функции спинного мозга
Строение и функции спинного мозга Звук, который не слышат взрослые
Звук, который не слышат взрослые Крокодилы и ящерицы
Крокодилы и ящерицы Жизненные формы растений
Жизненные формы растений 06 __________
06 __________ Птицы России. Викторина в 1 классе
Птицы России. Викторина в 1 классе Презентация на тему ЭВОЛЮЦИЯ ЧЕЛОВЕКА
Презентация на тему ЭВОЛЮЦИЯ ЧЕЛОВЕКА  Процесс кровообращения. Анатомия и физиология сердца
Процесс кровообращения. Анатомия и физиология сердца Карнавал бабочек
Карнавал бабочек Jean Batiste Lamarck
Jean Batiste Lamarck Пищеварительная система
Пищеварительная система Презентация на тему Неорганические вещества клетки
Презентация на тему Неорганические вещества клетки  Презентация на тему "Понятие о регенерации. Эволюция способности к регенерации у позвоночных" - презентации по Биологии
Презентация на тему "Понятие о регенерации. Эволюция способности к регенерации у позвоночных" - презентации по Биологии Покрытосеменные (цветковые) растения
Покрытосеменные (цветковые) растения