- Легочная эмфизема семейная у детей

Содержание
- 2. Эта болезнь (недостаточность альфа-1-антитрипсина — 1-АТ) является наследственной по аутосомно-рецессивному типу наследования. Среди детей, больных неспецифическими
- 4. При таком дефиците протеаз (химотрипсин, трипсин, нейтральная протеаза, эластаза) циркулирующие гранулоциты и моноциты разрушают ткань легких,
- 5. Экзогенные факторы, в частности продукты курения, воздушные полютанты, ксенобиотики и другие оксиданты, способны нарушать про-теафзно-антипротеазное равновесие
- 6. Клиника, диагностика Основной жалобой больных является одышка, возникающая вначале при значительных, а затем при все более
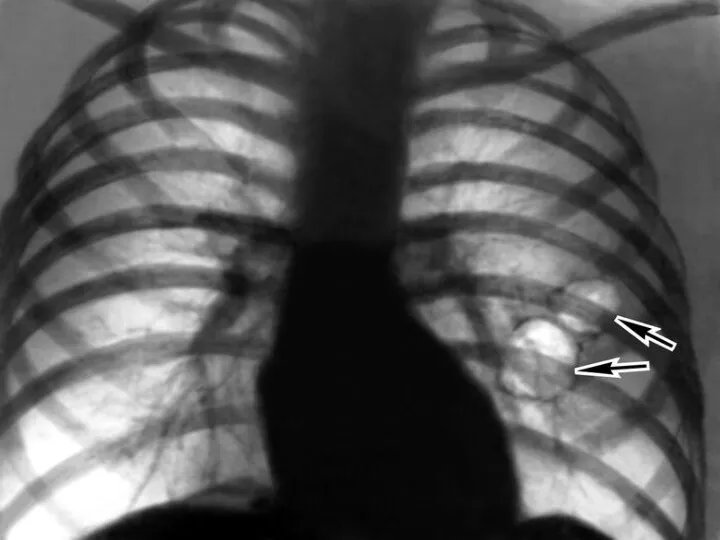
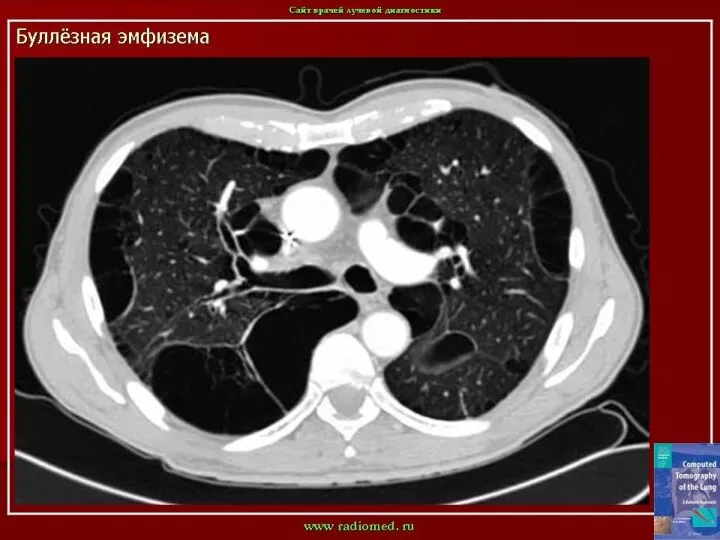
- 7. Рентгенологически обнаруживается увеличение прозрачности легочных полей, в типичных случаях более выраженное в нижних отделах легких, где
- 10. Лечение До недавнего времени лечение врожденного дефицита а-1-ингибиторов протеаз считалось малоперспективным. Заместительная терапия может осуществляться с
- 11. Прогноз Прогноз заболевания достаточно серьезный, так как наиболее эффективные методы терапии до настоящего времени не разработаны.
- 13. Скачать презентацию
Слайд 2Эта болезнь (недостаточность альфа-1-антитрипсина — 1-АТ) является наследственной по аутосомно-рецессивному типу наследования.
Среди
Эта болезнь (недостаточность альфа-1-антитрипсина — 1-АТ) является наследственной по аутосомно-рецессивному типу наследования.
Среди

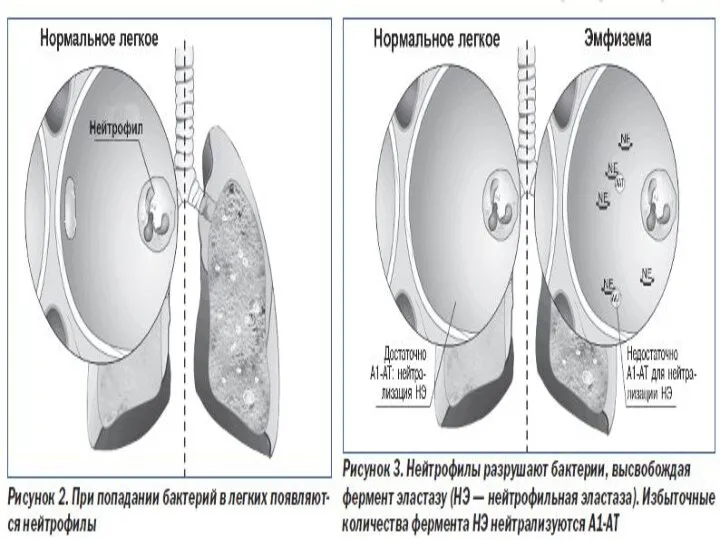
Слайд 4При таком дефиците протеаз (химотрипсин, трипсин, нейтральная протеаза, эластаза) циркулирующие гранулоциты и
При таком дефиците протеаз (химотрипсин, трипсин, нейтральная протеаза, эластаза) циркулирующие гранулоциты и

Слайд 5Экзогенные факторы, в частности продукты курения, воздушные полютанты, ксенобиотики и другие оксиданты,
Экзогенные факторы, в частности продукты курения, воздушные полютанты, ксенобиотики и другие оксиданты,

Слайд 6Клиника, диагностика
Основной жалобой больных является одышка, возникающая вначале при значительных, а затем
Клиника, диагностика
Основной жалобой больных является одышка, возникающая вначале при значительных, а затем

Слайд 7Рентгенологически обнаруживается увеличение прозрачности легочных полей, в типичных случаях более выраженное в
Рентгенологически обнаруживается увеличение прозрачности легочных полей, в типичных случаях более выраженное в

Слайд 10Лечение
До недавнего времени лечение врожденного дефицита а-1-ингибиторов протеаз считалось малоперспективным. Заместительная терапия
Лечение
До недавнего времени лечение врожденного дефицита а-1-ингибиторов протеаз считалось малоперспективным. Заместительная терапия

Слайд 11Прогноз
Прогноз заболевания достаточно серьезный, так как наиболее эффективные методы терапии до настоящего
Прогноз
Прогноз заболевания достаточно серьезный, так как наиболее эффективные методы терапии до настоящего

 №4 Проблемы смерти и умирания в биоэтике. Эвтаназия. Паллиативная медицина
№4 Проблемы смерти и умирания в биоэтике. Эвтаназия. Паллиативная медицина сп при норм теч бер
сп при норм теч бер проект рабочего места для мастера маникюра
проект рабочего места для мастера маникюра Советы доктора неболейка
Советы доктора неболейка Презентация 4
Презентация 4 Алдын ала жоспарланған және халықаралық егу сертификатын талап ететін жұқпалар
Алдын ала жоспарланған және халықаралық егу сертификатын талап ететін жұқпалар Правила наложения повязок
Правила наложения повязок Неотложные состояния в урологии
Неотложные состояния в урологии Вирусные пневмонии
Вирусные пневмонии Кафедра медицины катастроф
Кафедра медицины катастроф Нарушения гемостаза у детей
Нарушения гемостаза у детей Хронический гепатит
Хронический гепатит Пиодермия.ДильдораИсаевна
Пиодермия.ДильдораИсаевна Вегетативная нервная система
Вегетативная нервная система Профилактика заболеваний
Профилактика заболеваний Қан ауруларымен балалардын реабилитация. Темір жетіспеушілік анемия
Қан ауруларымен балалардын реабилитация. Темір жетіспеушілік анемия Экстракардиальные и интракардиальные предшественики для регенерации миокарда
Экстракардиальные и интракардиальные предшественики для регенерации миокарда Удаленные предрейсовые и предсменные медицинские осмотры
Удаленные предрейсовые и предсменные медицинские осмотры Опрелость. Пролежни. Профилактика опрелостей и пролежней
Опрелость. Пролежни. Профилактика опрелостей и пролежней Здоровье человека и продолжительность жизни
Здоровье человека и продолжительность жизни Клиническая симптоматология острой ревматической лихорадки (ОРЛ)
Клиническая симптоматология острой ревматической лихорадки (ОРЛ) Diagnostyka rodzaju skóry (тест)
Diagnostyka rodzaju skóry (тест) Центр медицинского массажа и косметологии
Центр медицинского массажа и косметологии Научно-практический календарь травматологии и ортопедии
Научно-практический календарь травматологии и ортопедии Особенности лабораторной диагностики кожного мастоцитоза у детей
Особенности лабораторной диагностики кожного мастоцитоза у детей Здоровый образ жизни
Здоровый образ жизни Ушная раковина и телосложение девушек 18-19 лет: изменчивость и связи
Ушная раковина и телосложение девушек 18-19 лет: изменчивость и связи Группа оздоровительной направленности с установленным диагнозом Сахарный диабет I типа
Группа оздоровительной направленности с установленным диагнозом Сахарный диабет I типа