- Синдром Ангельмана. Синдром счастливой куклы

Содержание
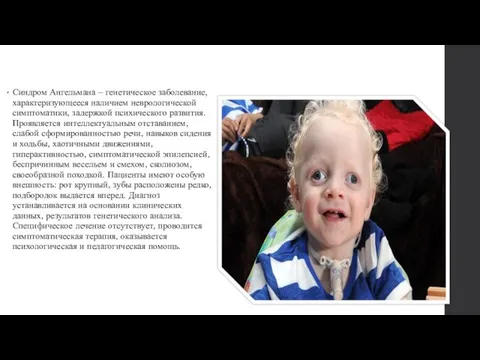
- 2. Синдром Ангельмана – генетическое заболевание, характеризующееся наличием неврологической симптоматики, задержкой психического развития. Проявляется интеллектуальным отставанием, слабой
- 3. Причины Факторы развития синдрома Ангельмана продолжают исследоваться. Генетический дефект обнаружен в 15 хромосоме материнского набора, но
- 4. Патеногенез Основа синдрома Ангельмана – нарушение функций гена UBE3A, расположенного в пятнадцатой материнской хромосоме. Этот ген
- 5. Симптомы Клинически заболевание проявляется в возрасте от 6 до 12 месяцев. Постепенно нарастает задержка развития, ранее
- 6. Диагностика Обследованием детей с подозрением на синдром Ангельмана занимаются врачи-неврологи, психиатры и генетики. Родители предъявляют жалобы
- 8. Скачать презентацию
Слайд 2Синдром Ангельмана – генетическое заболевание, характеризующееся наличием неврологической симптоматики, задержкой психического развития.
Синдром Ангельмана – генетическое заболевание, характеризующееся наличием неврологической симптоматики, задержкой психического развития.
Слайд 3Причины
Факторы развития синдрома Ангельмана продолжают исследоваться. Генетический дефект обнаружен в 15 хромосоме
Причины
Факторы развития синдрома Ангельмана продолжают исследоваться. Генетический дефект обнаружен в 15 хромосоме

Слайд 4Патеногенез
Основа синдрома Ангельмана – нарушение функций гена UBE3A, расположенного в пятнадцатой материнской
Патеногенез
Основа синдрома Ангельмана – нарушение функций гена UBE3A, расположенного в пятнадцатой материнской

Слайд 5Симптомы
Клинически заболевание проявляется в возрасте от 6 до 12 месяцев. Постепенно
Симптомы
Клинически заболевание проявляется в возрасте от 6 до 12 месяцев. Постепенно

Слайд 6Диагностика
Обследованием детей с подозрением на синдром Ангельмана занимаются врачи-неврологи, психиатры и
Диагностика
Обследованием детей с подозрением на синдром Ангельмана занимаются врачи-неврологи, психиатры и

 Кровотечения в последовом и раннем послеродовом периодах
Кровотечения в последовом и раннем послеродовом периодах Өндірістік шаң әсерінен тәжірибелік жануарлардың өкпе ұлпасындағы морфологиялық өзгерістер
Өндірістік шаң әсерінен тәжірибелік жануарлардың өкпе ұлпасындағы морфологиялық өзгерістер Pcos diet and lifestyle
Pcos diet and lifestyle Внеурочная деятельность.Этика - азбука добра. 3 класс
Внеурочная деятельность.Этика - азбука добра. 3 класс Жедел ішек өтімсіздігі
Жедел ішек өтімсіздігі Острый живот в акушерстве – острая хирургическая патология у беременных
Острый живот в акушерстве – острая хирургическая патология у беременных Современные стандарты ведения пациентов с ОНМК
Современные стандарты ведения пациентов с ОНМК Минимально инвазивная эзофагэктомия: беспристрастный разговор
Минимально инвазивная эзофагэктомия: беспристрастный разговор 2022 Дифф. диагноз заболеваний суставов у детей (1)
2022 Дифф. диагноз заболеваний суставов у детей (1) Chronic gastritis
Chronic gastritis Лимфа ағысын анықтайтын факторлар. Лимфа жүйесін зерттеудің заманауи әдістері
Лимфа ағысын анықтайтын факторлар. Лимфа жүйесін зерттеудің заманауи әдістері Дыхание - это жизнь
Дыхание - это жизнь 2020 – год медицинской сестры и акушерки (ВОЗ)
2020 – год медицинской сестры и акушерки (ВОЗ) Современные технологии преаналитического этапа исследования газов и электролитов крови
Современные технологии преаналитического этапа исследования газов и электролитов крови Основные проблемы медицины социальной сферы
Основные проблемы медицины социальной сферы Планирование и моделирование костно-реконструктивных операций и зубной имплантации
Планирование и моделирование костно-реконструктивных операций и зубной имплантации Тіндер мен ағзалар электрографисының биофизикалық негіздері
Тіндер мен ағзалар электрографисының биофизикалық негіздері FAST протокол
FAST протокол ALPPS: тайна пяти букв
ALPPS: тайна пяти букв Принципы лечения хирургической инфекции
Принципы лечения хирургической инфекции Гигие́на (греч. ὑγιεινός здоровый)
Гигие́на (греч. ὑγιεινός здоровый) Функциональная анатомия мочевыделительной системы
Функциональная анатомия мочевыделительной системы Чрескостный остеосинтез верхних конечностей
Чрескостный остеосинтез верхних конечностей Індикатор втоми
Індикатор втоми Клуб Здоровье
Клуб Здоровье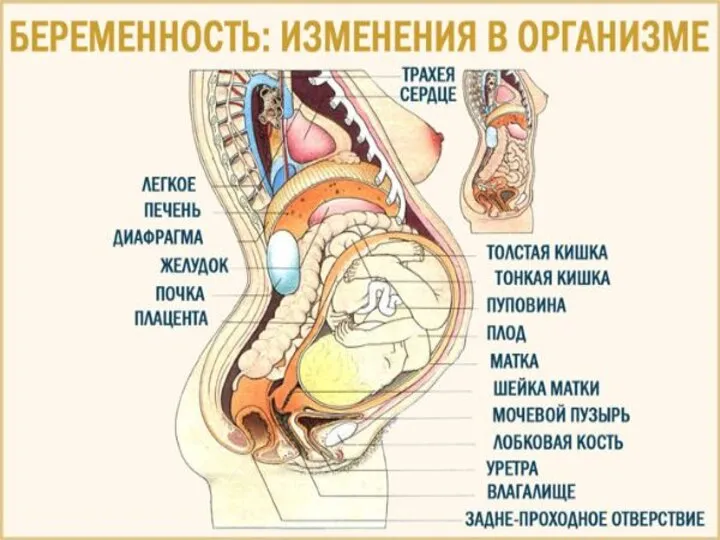 04 Физиол изм при бер в орг-ме женщины
04 Физиол изм при бер в орг-ме женщины 92500 (4)
92500 (4) Ранний период беременности
Ранний период беременности