- Anemia in children

Содержание
- 3. Аnemia – pathologic state, accompanied by decrease in the level of hemoglobin and the quantity of
- 4. Anemia is defined as hematocrit (Hct), hemoglobin (Hb), red blood cells (RBC) concentration > 2 SD
- 5. Anemia is a condition in which a person’s blood has a lower number of red blood
- 6. Normal red blood cells live about 120 days in the bloodstream and then die. Their main
- 7. Erythrocytes - less informative index of anemia than the level of hemoglobin. In the general practice
- 8. RETICULOCYTE Reticulocytes are immature red blood cells, typically composing about 1% of the red cells in
- 9. The clinical index of blood. (Institute of Pediatric Hematology, the drafters of the A.G .Rumyantsev, E.B
- 11. Normal levels of index in CBT in children different ages.
- 13. Anemia criteria (capillary blood) in newborns In newborns of the first and the second weeks of
- 14. Normal index of Hb according WHO criteria (capillary blood) : In infants from 2 to 6
- 15. CAUSES OF NEONATAL ANEMIA: BLOOD LOSS (POSTHEMORRHAGIC) the commonest cause of anemia, including: A. Obstetrical causes:
- 16. C. Feto-placental transfusion Occurs only with monochorionic (i.e., monozygotic) twins and if placental vessels allow shunting
- 17. Blood loss Gastrointestinal: gastro-oesophageal reflux, Meckel’s diverticulum, cow’s milk protein intolerance; Parasites – hookworm; Menstruation in
- 18. 2. INCREASED RBC DESTRUCTION (HAEMOLYTIC): Endogenic causes: Hereditary RBC disorders (rare), including: •RBC Enzyme defects (e.g.,
- 19. B. Exogenic causes: • Immune hemolysis: -Rh incompatibility, - ABO incompitability, - Minor blood group incompatibility
- 20. 3. DECREASED RBC PRODUCTION (HYPOPLASTIC, DEFICIENCY): A. Anemia of prematurity due to transient deficiency of erythropoietin;
- 21. Physiological anemia of infancy Normal newborn: - High Hb level progressively declines by 8-12 week of
- 22. I. Anemia resulting from blood loss: A) Acute; B) Chronical. II. Anemia resulting from a insufficiency
- 23. B) Acquired aplastic anemia (atrophic anemia, anemia gravis): a) With Pancytopenia (acute, subacute, chronic); b) With
- 24. b) microcytic anemia: - iron deficiency (asiderotic anemia); copper deficiency; Lead poisoning; Thalassaemia. F) physiologic anemia
- 25. A) Aquired: A. Immunopathologic; Б. Infectious; B. Vitamin deficiency; Г. Toxic; Д. Marchiafava-Micheli anemia; Е. Disseminated
- 26. IV. Mixed genesis anemia: A. In acute infections, sepsis. Б. In burns. B. In tumors and
- 27. Anemia are divided in according to number of reticulocyte : Regenerative- reticulocyte from 1.5 to 5%
- 28. Anemia are divided in according erythrocytic indexes microcytic anemia (MCV less than 75 fl); normocytic anemia
- 29. Classification of anemia
- 30. Anemia are divided in order of severity
- 31. Morphology Nomenclature of Erythrocytes Acanthocyte Dacrocyte Drepanocyte Echinocyte Elliptocyte Megalocyte Schizocyte Spherocyte Stomatocyte Spicule: Liver disease
- 32. Anemia are divided in according Colour Index Normochromal anemia – colour index = 0,85 - 1,05;
- 33. IRON DEFICIENCY ANEMIA (IDA) IDА is recorded in 20% of the world's population. 83-90% of all
- 34. CAUSES FOR DEVELOPMENT of IDA Alimentary iron deficiency as a consequence of an unbalanced diet; Increasing
- 35. They are: - 75-80% belongs to the hemoglobin; - 20 - 25% reserve; - 5-10% part
- 36. Anemic Syndrome - Decrease amount of Hemoglobin Complaints: General weakness, reduction in the appetite, physical and
- 37. Sideropenic Syndrome (Deficit of Iron) dystrophic changes in the skin and its appendages (shedding of hair,
- 38. Laboratory Signs of Iron Deficit Anemia A decrease MCV - less than 75; Reduction in the
- 39. Developmental Stages of Iron Deficiency Anemia (WHO, 1977) Pre-latent (exhaustion of iron reserve in tissues; index
- 40. Differential Diagnosis of Iron Deficiency Anemia it is carried out with other forms of the hypochromic
- 41. Ferritin Water-soluble complex of iron hydroxide with the protein apoferritin. It is located in cells of
- 42. Serum Transferrin (Beta-globulin). Main function to transport of absorbed iron in the depot (liver, spleen), into
- 43. Transferrin
- 44. – We take in 10-30mg Fe/day; – We absorb 0.5-1.0mg/day (5.5-10%); – Absorption may increase to
- 45. Treatment of Iron Deficiency Anemia Diet: meat, liver, yeast, fish. Oral preparations: the rate of absorbtion
- 46. During first 3 days - half dose of the selected active substance. Possibilities: dark colour of
- 47. Therapeutic dose of Iron drug 0 – 3 years – 5 – 8 mg/kg/day; 3 –
- 48. Indication for Parenteral Introduction of Iron in exceptional cases; in severe iron deficiency anemia; intolerance of
- 49. Complications of Parenteral Introduction Local reactions (pains, phlebitis); General reactions (anaphylaxis, fever, head and articulate pains,
- 50. Signs of overdose of Iron In the first 6-8 hours - epigastral pains, nausea, vomiting (including
- 51. Iron Overload Syndrome ! Human does not have special mechanism of the excretion of iron! Its
- 52. Megaloblastic anemia it is inheritable and acquired anemia when in bone marrow the megaloblasts are present.
- 53. Causes of B12 Deficiency Anemia Decreased ingestion (e.g., poor dietary intake); Atrophy of the mucous membrane
- 54. Causes of Folic Acid Deficiency Alimentary; Increased need (prematurity birth, rapid growth rate, pregnancy); Feeding by
- 55. Clinical Anemic syndrome; Skin is pale with the lemon shade; Slight jaundice of the scleras; Glossitis,
- 56. Signs only for B12-Deficiency Anemia Damage of CNS - funicular myelosis (degeneration and the sclerosis of
- 57. Diagnosis of B12 and Folic acid Deficiency Anemia
- 58. Bone marrow: Irritation in erythroid lineage, megaloblasts, the disintegration of erithrokaryocytes. Biochemical Analysis of Blood an
- 60. Treatment of megaloblastic anemia Treatment of megaloblastic anemia depends on the underlying cause. Folate deficiency due
- 61. Oral B-12 supplementation is as effective as parenteral supplementation in patients with nutritional deficiency. Even in
- 62. Criteria of Effective Treatment Subjective improvement of patient condition during the first days of treatment; Reticulocytosis,
- 63. Folic acid 0-6 months: 65 mcg/day PO 7-12 months: 80 mcg/day PO 1-4 years: 150 mcg/day
- 64. Hemolytic anemia = reduced red-cell life span. Hemolysis is the premature destruction of erythrocytes. A hemolytic
- 65. Intravascular hemolysis occurs in hemolytic anemia due to the following: - Artificial cardiac valves; - Glucose-6-phosphate
- 66. Laboratory sings of Intravascular hemolysis: Indirect hyperbilirubinemia; Erythroid hyperplasia; Hemoglobinemia; Reticulocytosis; Hemoglobinuria; Absence or reduced of
- 67. Extravascular hemolysis: Red cells destruction occurs in reticuloendothelial system; Clinical states associated with extravascular hemolysis: autoimmune
- 68. Laboratory sings of Extravascular hemolysis: Indirect hyperbilirubinemia; Erythroid hyperplasia; Inareased excretion of bilirubin by bile; Hemosiderosis.
- 69. Etiology Hereditary disorders may cause hemolysis as a result of erythrocyte membrane abnormalities, enzymatic defects, and
- 70. Clinical features of hemolytic anemia: Patients with minimal or long-standing hemolytic anemia may be asymptomatic, and
- 71. Bronze skin color and diabetes occur in hematosiderosis; iron overload may occur in patients who have
- 72. Laboratory sings of hemolytic anemia: normocytic/macrocytic, hyperchromatic anemia; reticulocytosis; Increased serum iron; Antiglobin Coomb’ test is
- 73. Autoimmune hemolytic anemia it is status when in organism development antibodies against own erythrocytes with unchanged
- 74. Autoimmune hemolytic anemia caused by warm-reactive antibodies: Primary. Secondary: 1. acute - viral infections; - drugs
- 75. Autoimmune hemolytic anemia caused by cold-reactive antibodies: Primary. Secondary: - mycoplasma infections; - viral infections; -
- 76. Autoimmune hemolytic anemia - diagnosis - Possitive direct Coombs’ test. Treatment: Steroids; Splenectomy; Immunosupressive agents; Transfusion.
- 77. Hereditary microspherocytosis 1. Pathophysiology: red cell membrane protein defects (spectrin deficiency) resulting cytoskeleton instability. 2. Family
- 78. Thalassemia syndromes a heterogeneous group of inherited anemia characterized by reduced or absent synthesis of either
- 79. Alpha Thalassemia The alpha thalassemia patient's hemoglobin does not produce enough alpha protein. This type is
- 80. One faulty (mutated) gene - there are either no symptoms at all, or they are very
- 81. Three mutated genes - the patient will have hemoglobin H disease, i.e. chronic anemia. A person
- 82. Mortality/Morbidity α thalassemia major is a mortal disease, and virtually all affected fetuses are born with
- 83. Beta Thalassemia We need two globin genes to make beta globin chains. We get one from
- 84. Severity of beta thalassemia also depends on how many genes are mutated: If one globin gene
- 85. Signs And Symptoms Of Beta Thalassemia The majority of infants with beta thalassemia will not have
- 86. In patients with various types of β thalassemia, mortality and morbidity vary according to the severity
- 87. Age Despite thalassemia's inherited nature, age at onset of symptoms varies significantly. In α thalassemia, clinical
- 88. People with thalassemia mainly have anemia-like symptoms. Jaundice; Fatigue; Pale skin; Cold hands and feet; Dyspnea;
- 89. Signs And Symptoms Of Alpha Thalassemia The majority of children with hemoglobin H are generally healthy.
- 90. Diagnosing of thalassemia A complete blood count (CBC) - to measure hemoglobin levels, quantities of red
- 91. The WBC count is usually elevated in β thalassemia major; this is due, in part, to
- 92. Treatment of thalassemia Blood transfusions - this is done to replenish hemoglobin and red blood cell
- 93. Complications of thalassemia Iron overload; Enlarged spleen (splenomegaly); Infection; Bone deformities.
- 94. Congenital sideroblastic anemias Congenital sideroblastic anemias generally present with lower hemoglobin and more microcytosis than myelodysplastic
- 95. Etiology Congenital causes of sideroblastic anemia include the following: δ-ALAS mutation; ABC7 mutation; PSU1 mutation; Pearson
- 96. Major causes of death in cases of sideroblastic anemia are secondary hemochromatosis from transfusions and leukemia.
- 97. Clinical Presentation Incoordination (cerebellar symptoms); Failure of growth; Diarrhea (malabsorption); Polyuria, blindness, deafness (associated with DIDMOAD
- 98. Medication history of antibiotics, antituberculous agents, chelators, or chemotherapy; Ingestion of supplements, especially zinc; Prolonged dependence
- 99. Physical Examination General - Growth retardation in children; Vital signs – Hypothermia; Oral - Lead line
- 100. Complete Blood Cell Count and Peripheral Smear CBC count usually reveals anemia. The mean corpuscular volume
- 101. Sideroblastic anemia also seen in combined vitamin B-12 deficiency with iron deficiency. But iron studies may
- 102. Treatment Treatment of sideroblastic anemia may include removal of toxic agents; administration of pyridoxine, thiamine, or
- 103. SICKLE CELL ANEMIA A serious condition in which red blood cells can become sickle-shaped. In sickle
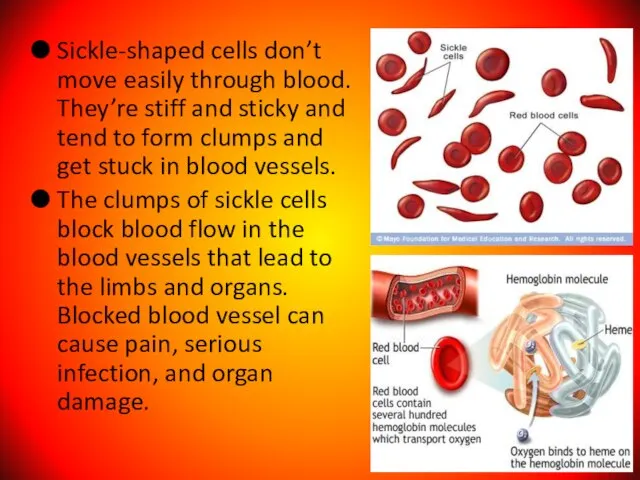
- 104. Sickle-shaped cells don’t move easily through blood. They’re stiff and sticky and tend to form clumps
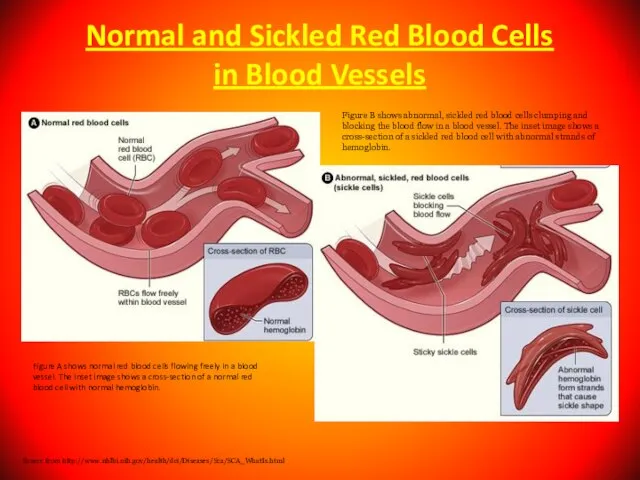
- 105. Normal and Sickled Red Blood Cells in Blood Vessels Figure A shows normal red blood cells
- 106. People who have sickle cell anemia are born with it; means inherited, lifelong condition. They inherit
- 107. Inheritance of Sickle Cell Anemia If one parent has sickle cell anaemia (HbSS) and the other
- 108. Inheritance of Sickle Cell Anemia If both parents have sickle cell trait (HbAS) there is a
- 110. Signs and Symptoms Individual signs and symptoms varies. Some have mild symptoms, others have very severe
- 111. Differential Diagnoses Acute Anemia; Carotid Cavernous Fistula; Hands Rheumatoid Arthritis; Hemoglobin C Disease; Hemolytic Anemia; Legg-Calve-Perthes
- 112. Complication of Sickle Cell Anemia Splenic Crisis Infections Acute Chest Syndrome Delayed growth and puberty in
- 113. Treatment Effective treatment is available to help relieve the symptoms and complications of sickle cell anemia.no
- 114. Prevention Identify what can trigger the “Crisis” such as stress, avoid extremes of heat and cold
- 115. Aplastic Anemia is a syndrome of bone marrow failure characterized by peripheral pancytopenia and marrow hypoplasia.
- 116. Fanconi Anemia Fanconi anemia is the most frequently reported of the rare inherited bone marrow failure
- 117. Multiple congenital anomalies (60-75%): Short stature, petechiae and bruises, abnormal skin pigmentation, café au lait spots,
- 118. A 3-year-old patient with Fanconi anemia. Note the multiple birth defects, including short stature, microcephaly, microphthalmia,
- 119. Bone marrow failure: Thrombocytopenia, leukopenia, or aplastic anemia; most patients with Fanconi anemia have bone marrow
- 120. Gonads may display the following abnormalities: Males - Hypogenitalia, undescended testes, hypospadias, abnormal or absent testis,
- 121. Cancer: Hematologic malignancies are common with Fanconi anemia and myelodysplasia: acute myeloid leukemia (AML) being the
- 122. Supportive Care Treatment is recommended for significant cytopenias, such as hemoglobin less than 8 g/dL, platelets
- 124. Скачать презентацию
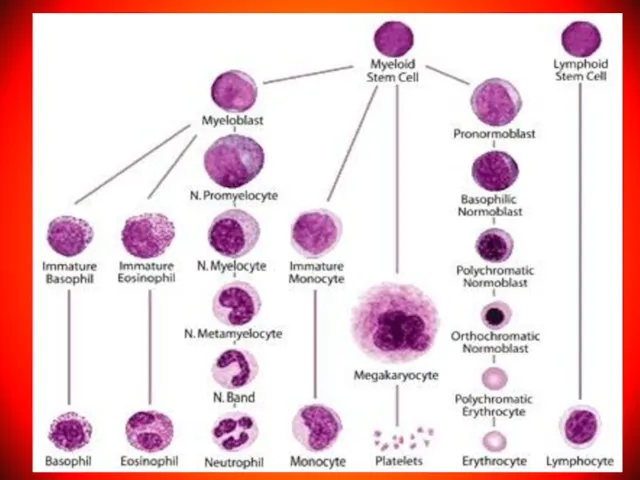
Слайд 3Аnemia – pathologic state, accompanied by decrease in the level of hemoglobin
Аnemia – pathologic state, accompanied by decrease in the level of hemoglobin

Слайд 4 Anemia is defined as hematocrit (Hct), hemoglobin (Hb), red blood cells
Anemia is defined as hematocrit (Hct), hemoglobin (Hb), red blood cells

Слайд 5Anemia
is a condition in which a person’s blood has a lower number
Anemia
is a condition in which a person’s blood has a lower number

Слайд 6Normal red blood cells live about 120 days in the bloodstream and
Normal red blood cells live about 120 days in the bloodstream and

Слайд 7 Erythrocytes
- less informative index of anemia than the level of hemoglobin.
Erythrocytes
- less informative index of anemia than the level of hemoglobin.

Слайд 8
RETICULOCYTE
Reticulocytes are immature red blood cells, typically composing about 1% of the
RETICULOCYTE
Reticulocytes are immature red blood cells, typically composing about 1% of the

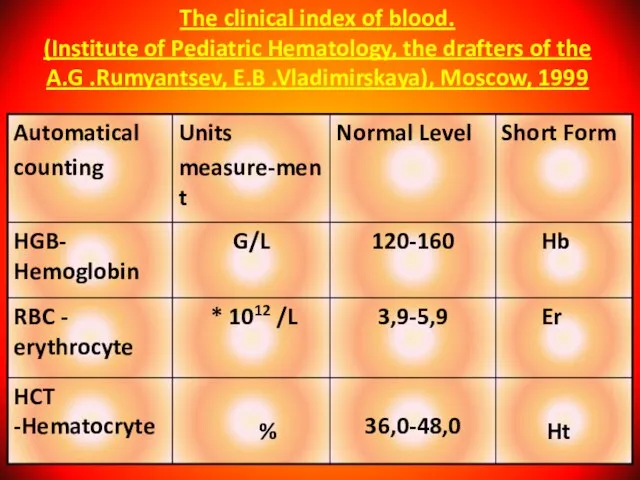
Слайд 9The clinical index of blood.
(Institute of Pediatric Hematology, the drafters of
The clinical index of blood. (Institute of Pediatric Hematology, the drafters of

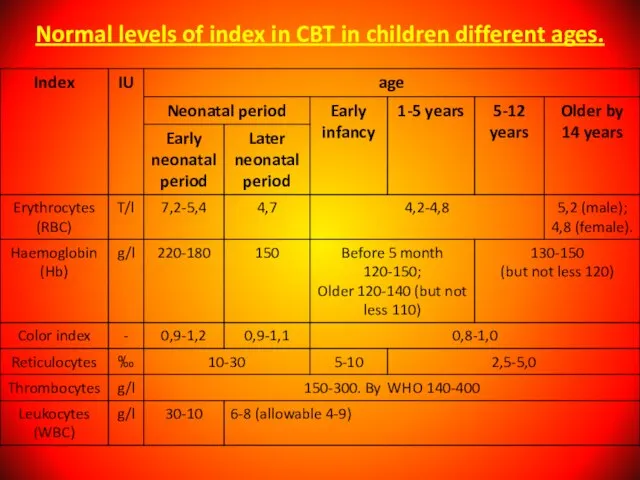
Слайд 11Normal levels of index in CBT in children different ages.
Normal levels of index in CBT in children different ages.
Слайд 13Anemia criteria (capillary blood) in newborns
In newborns of the first and
Anemia criteria (capillary blood) in newborns
In newborns of the first and

Слайд 14 Normal index of Hb according WHO criteria (capillary blood) :
In infants
Normal index of Hb according WHO criteria (capillary blood) :
In infants

Слайд 15CAUSES OF NEONATAL ANEMIA:
BLOOD LOSS (POSTHEMORRHAGIC)
the commonest cause of anemia,
CAUSES OF NEONATAL ANEMIA:
BLOOD LOSS (POSTHEMORRHAGIC)
the commonest cause of anemia,

Слайд 16C. Feto-placental transfusion Occurs only with monochorionic (i.e., monozygotic) twins and if
C. Feto-placental transfusion Occurs only with monochorionic (i.e., monozygotic) twins and if

Слайд 17Blood loss
Gastrointestinal:
gastro-oesophageal reflux,
Meckel’s diverticulum,
cow’s milk protein intolerance;
Parasites – hookworm;
Menstruation in
Blood loss
Gastrointestinal:
gastro-oesophageal reflux,
Meckel’s diverticulum,
cow’s milk protein intolerance;
Parasites – hookworm;
Menstruation in

Слайд 182. INCREASED RBC DESTRUCTION (HAEMOLYTIC):
Endogenic causes:
Hereditary RBC disorders (rare), including:
•RBC Enzyme
2. INCREASED RBC DESTRUCTION (HAEMOLYTIC):
Endogenic causes:
Hereditary RBC disorders (rare), including:
•RBC Enzyme

Слайд 19B. Exogenic causes:
• Immune hemolysis: -Rh incompatibility,
- ABO incompitability,
- Minor
B. Exogenic causes:
• Immune hemolysis: -Rh incompatibility,
- ABO incompitability,
- Minor

Слайд 203. DECREASED RBC PRODUCTION (HYPOPLASTIC, DEFICIENCY):
A. Anemia of prematurity due to transient
3. DECREASED RBC PRODUCTION (HYPOPLASTIC, DEFICIENCY):
A. Anemia of prematurity due to transient

Слайд 21Physiological anemia of infancy
Normal newborn:
- High Hb level progressively declines by 8-12
Physiological anemia of infancy
Normal newborn:
- High Hb level progressively declines by 8-12

Слайд 22 I. Anemia resulting from blood loss:
A) Acute;
B) Chronical.
II. Anemia resulting from
I. Anemia resulting from blood loss:
A) Acute;
B) Chronical.
II. Anemia resulting from

Слайд 23B) Acquired aplastic anemia (atrophic anemia, anemia gravis):
a) With Pancytopenia (acute, subacute,
B) Acquired aplastic anemia (atrophic anemia, anemia gravis):
a) With Pancytopenia (acute, subacute,

Слайд 24 b) microcytic anemia:
- iron deficiency (asiderotic anemia);
copper deficiency;
Lead poisoning;
b) microcytic anemia:
- iron deficiency (asiderotic anemia);
copper deficiency;
Lead poisoning;

Слайд 25 A) Aquired:
A. Immunopathologic;
Б. Infectious;
B. Vitamin deficiency;
Г. Toxic;
Д.
A) Aquired:
A. Immunopathologic;
Б. Infectious;
B. Vitamin deficiency;
Г. Toxic;
Д.

Слайд 26IV. Mixed genesis anemia:
A. In acute infections, sepsis.
Б. In burns.
B. In
IV. Mixed genesis anemia:
A. In acute infections, sepsis.
Б. In burns.
B. In

Слайд 27Anemia are divided in according to number of reticulocyte :
Regenerative- reticulocyte
Anemia are divided in according to number of reticulocyte :
Regenerative- reticulocyte

Слайд 28Anemia are divided in according erythrocytic indexes
microcytic anemia (MCV less than
Anemia are divided in according erythrocytic indexes
microcytic anemia (MCV less than

Слайд 29Classification of anemia
Classification of anemia
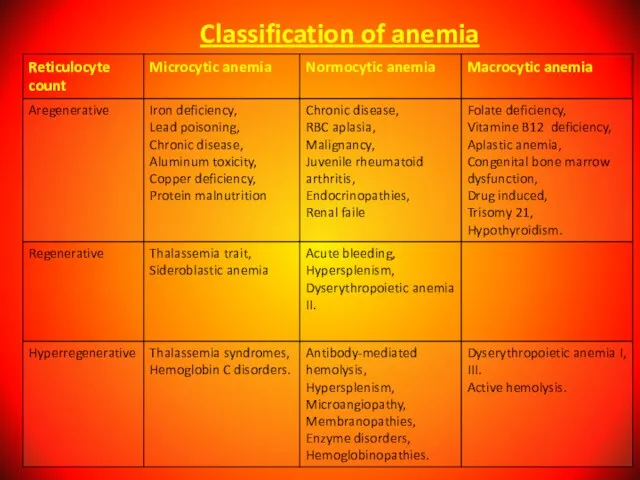
Слайд 30Anemia are divided in order of severity
Anemia are divided in order of severity

Слайд 31Morphology Nomenclature of Erythrocytes
Acanthocyte
Dacrocyte
Drepanocyte
Echinocyte
Elliptocyte
Megalocyte
Schizocyte
Spherocyte
Stomatocyte
Spicule: Liver disease
Tear: Thalassemia, Heinz body
Sickle: Sickle cell
Morphology Nomenclature of Erythrocytes
Acanthocyte
Dacrocyte
Drepanocyte
Echinocyte
Elliptocyte
Megalocyte
Schizocyte
Spherocyte
Stomatocyte
Spicule: Liver disease
Tear: Thalassemia, Heinz body
Sickle: Sickle cell

Слайд 32Anemia are divided in according Colour Index
Normochromal anemia – colour index
Anemia are divided in according Colour Index
Normochromal anemia – colour index

Слайд 33IRON DEFICIENCY ANEMIA (IDA)
IDА is recorded in 20% of the world's population.
IRON DEFICIENCY ANEMIA (IDA)
IDА is recorded in 20% of the world's population.

Слайд 34CAUSES FOR DEVELOPMENT of IDA
Alimentary iron deficiency as a consequence of an
CAUSES FOR DEVELOPMENT of IDA
Alimentary iron deficiency as a consequence of an

Слайд 35They are:
- 75-80% belongs to the hemoglobin;
- 20 - 25% reserve;
- 5-10%
They are: - 75-80% belongs to the hemoglobin; - 20 - 25% reserve; - 5-10%

Слайд 36Anemic Syndrome
- Decrease amount of Hemoglobin
Complaints: General weakness, reduction in the appetite,
physical
Anemic Syndrome
- Decrease amount of Hemoglobin
Complaints: General weakness, reduction in the appetite,
physical

Слайд 37Sideropenic Syndrome (Deficit of Iron)
dystrophic changes in the skin and its appendages
Sideropenic Syndrome (Deficit of Iron)
dystrophic changes in the skin and its appendages

Слайд 38Laboratory Signs of Iron Deficit Anemia
A decrease MCV - less than 75;
Reduction
Laboratory Signs of Iron Deficit Anemia
A decrease MCV - less than 75;
Reduction

Слайд 39Developmental Stages of Iron Deficiency Anemia (WHO, 1977)
Pre-latent (exhaustion of iron reserve
Developmental Stages of Iron Deficiency Anemia (WHO, 1977)
Pre-latent (exhaustion of iron reserve

Слайд 40Differential Diagnosis of Iron Deficiency Anemia
it is carried out with other forms
Differential Diagnosis of Iron Deficiency Anemia
it is carried out with other forms

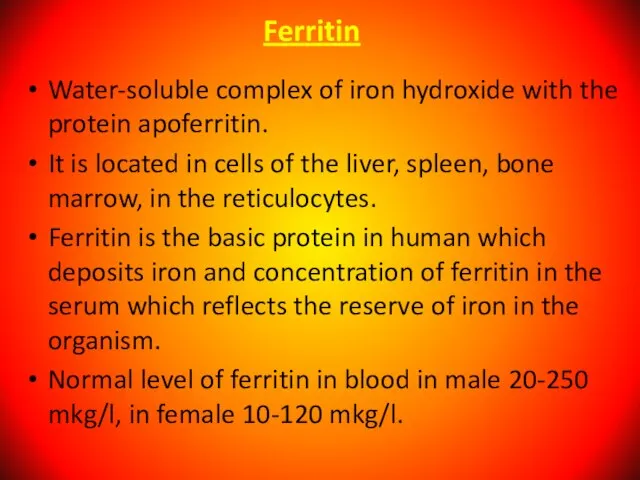
Слайд 41Ferritin
Water-soluble complex of iron hydroxide with the protein apoferritin.
It is located in
Ferritin
Water-soluble complex of iron hydroxide with the protein apoferritin.
It is located in
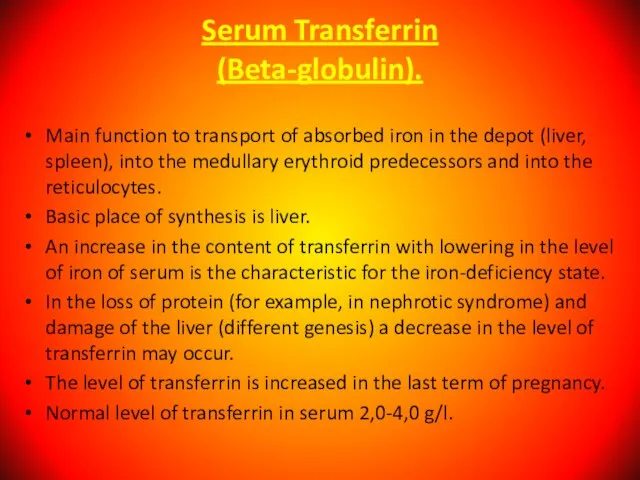
Слайд 42Serum Transferrin
(Beta-globulin).
Main function to transport of absorbed iron in the depot (liver,
Serum Transferrin
(Beta-globulin).
Main function to transport of absorbed iron in the depot (liver,
Слайд 43Transferrin
Transferrin

Слайд 44– We take in 10-30mg Fe/day;
– We absorb 0.5-1.0mg/day (5.5-10%);
– Absorption may
– We take in 10-30mg Fe/day;
– We absorb 0.5-1.0mg/day (5.5-10%);
– Absorption may

Слайд 45Treatment of Iron Deficiency Anemia
Diet: meat, liver, yeast, fish.
Oral preparations: the rate
Treatment of Iron Deficiency Anemia
Diet: meat, liver, yeast, fish.
Oral preparations: the rate

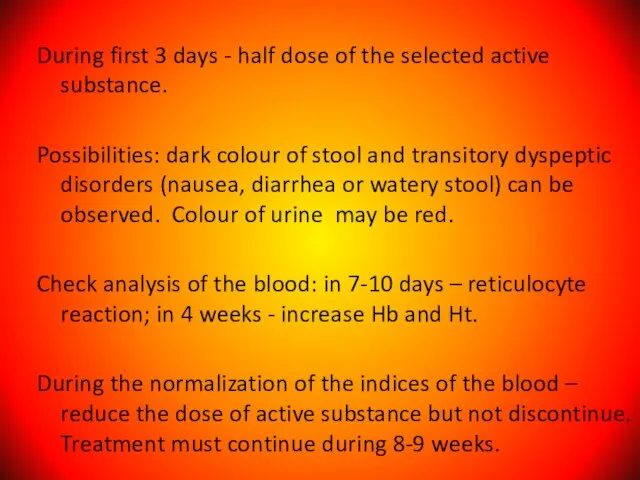
Слайд 46During first 3 days - half dose of the selected active substance.
During first 3 days - half dose of the selected active substance.
Слайд 47Therapeutic dose of Iron drug
0 – 3 years – 5 –
Therapeutic dose of Iron drug
0 – 3 years – 5 –

Слайд 48Indication for Parenteral Introduction of Iron
in exceptional cases;
in severe iron deficiency anemia;
intolerance
Indication for Parenteral Introduction of Iron
in exceptional cases;
in severe iron deficiency anemia;
intolerance

Слайд 49Complications of Parenteral Introduction
Local reactions (pains, phlebitis);
General reactions (anaphylaxis, fever, head and
Complications of Parenteral Introduction
Local reactions (pains, phlebitis);
General reactions (anaphylaxis, fever, head and

Слайд 50Signs of overdose of Iron
In the first 6-8 hours - epigastral pains,
Signs of overdose of Iron
In the first 6-8 hours - epigastral pains,

Слайд 51
Iron Overload Syndrome
! Human does not have special mechanism of the excretion
Iron Overload Syndrome
! Human does not have special mechanism of the excretion

Слайд 52Megaloblastic anemia it is inheritable and acquired anemia when in bone marrow
Megaloblastic anemia it is inheritable and acquired anemia when in bone marrow

Слайд 53Causes of B12 Deficiency Anemia
Decreased ingestion (e.g., poor dietary intake);
Atrophy of the
Causes of B12 Deficiency Anemia
Decreased ingestion (e.g., poor dietary intake);
Atrophy of the

Слайд 54Causes of Folic Acid Deficiency
Alimentary;
Increased need (prematurity birth, rapid growth rate, pregnancy);
Feeding
Causes of Folic Acid Deficiency
Alimentary;
Increased need (prematurity birth, rapid growth rate, pregnancy);
Feeding

Слайд 55Clinical
Anemic syndrome;
Skin is pale with the lemon shade;
Slight jaundice of the
Clinical
Anemic syndrome;
Skin is pale with the lemon shade;
Slight jaundice of the

Слайд 56Signs only for B12-Deficiency Anemia
Damage of CNS - funicular myelosis (degeneration and
Signs only for B12-Deficiency Anemia
Damage of CNS - funicular myelosis (degeneration and

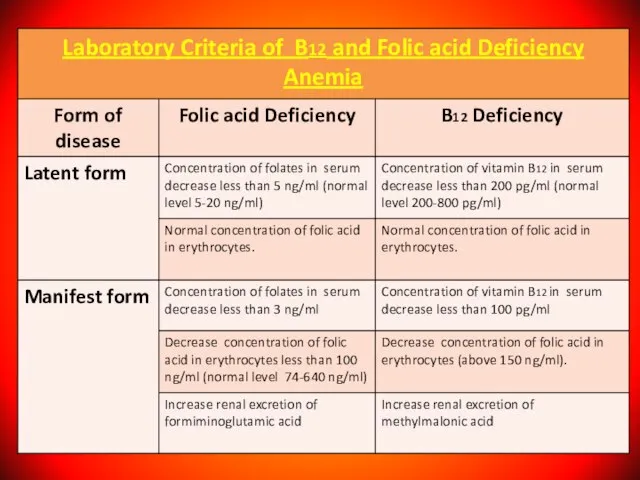
Слайд 57Diagnosis of B12 and Folic acid Deficiency Anemia
Diagnosis of B12 and Folic acid Deficiency Anemia

Слайд 58Bone marrow:
Irritation in erythroid lineage, megaloblasts,
the disintegration of erithrokaryocytes.
Biochemical Analysis of
Bone marrow:
Irritation in erythroid lineage, megaloblasts,
the disintegration of erithrokaryocytes.
Biochemical Analysis of

Слайд 60Treatment of megaloblastic anemia
Treatment of megaloblastic anemia depends on the underlying cause.
Treatment of megaloblastic anemia
Treatment of megaloblastic anemia depends on the underlying cause.

Слайд 61Oral B-12 supplementation is as effective as parenteral supplementation in patients with
Oral B-12 supplementation is as effective as parenteral supplementation in patients with

Слайд 62Criteria of Effective Treatment
Subjective improvement of patient condition during the first days
Criteria of Effective Treatment
Subjective improvement of patient condition during the first days

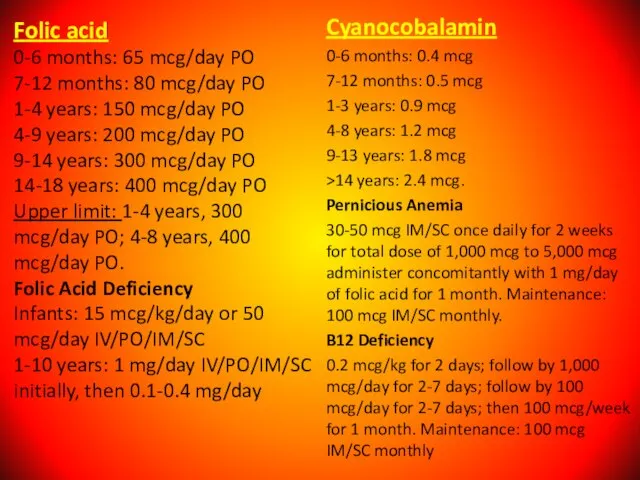
Слайд 63Folic acid
0-6 months: 65 mcg/day PO
7-12 months: 80 mcg/day PO
1-4 years: 150
Folic acid 0-6 months: 65 mcg/day PO 7-12 months: 80 mcg/day PO 1-4 years: 150
Слайд 64Hemolytic anemia = reduced red-cell
life span.
Hemolysis is the premature destruction of
Hemolytic anemia = reduced red-cell
life span.
Hemolysis is the premature destruction of

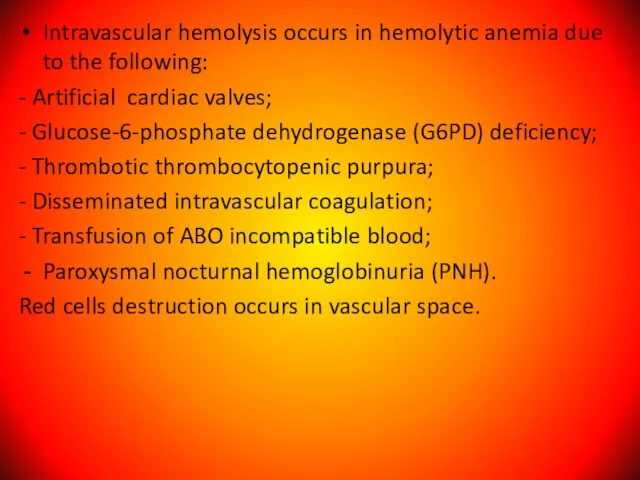
Слайд 65Intravascular hemolysis occurs in hemolytic anemia due to the following:
- Artificial
Intravascular hemolysis occurs in hemolytic anemia due to the following:
- Artificial
Слайд 66Laboratory sings of Intravascular hemolysis:
Indirect hyperbilirubinemia;
Erythroid hyperplasia;
Hemoglobinemia;
Reticulocytosis;
Hemoglobinuria;
Absence or reduced of
Laboratory sings of Intravascular hemolysis:
Indirect hyperbilirubinemia;
Erythroid hyperplasia;
Hemoglobinemia;
Reticulocytosis;
Hemoglobinuria;
Absence or reduced of

Слайд 67Extravascular hemolysis:
Red cells destruction occurs in reticuloendothelial system;
Clinical states associated with extravascular
Extravascular hemolysis:
Red cells destruction occurs in reticuloendothelial system;
Clinical states associated with extravascular

Слайд 68Laboratory sings of Extravascular hemolysis:
Indirect hyperbilirubinemia;
Erythroid hyperplasia;
Inareased excretion of bilirubin by
Laboratory sings of Extravascular hemolysis:
Indirect hyperbilirubinemia;
Erythroid hyperplasia;
Inareased excretion of bilirubin by

Слайд 69Etiology
Hereditary disorders may cause hemolysis as a result of erythrocyte membrane
Etiology
Hereditary disorders may cause hemolysis as a result of erythrocyte membrane

Слайд 70Clinical features of hemolytic anemia:
Patients with minimal or long-standing hemolytic anemia may
Clinical features of hemolytic anemia:
Patients with minimal or long-standing hemolytic anemia may

Слайд 71Bronze skin color and diabetes occur in hematosiderosis; iron overload may occur
Bronze skin color and diabetes occur in hematosiderosis; iron overload may occur

Слайд 72Laboratory sings of hemolytic anemia:
normocytic/macrocytic, hyperchromatic anemia;
reticulocytosis;
Increased serum iron;
Antiglobin Coomb’ test
Laboratory sings of hemolytic anemia:
normocytic/macrocytic, hyperchromatic anemia;
reticulocytosis;
Increased serum iron;
Antiglobin Coomb’ test

Слайд 73Autoimmune hemolytic anemia
it is status when in organism development antibodies against own
Autoimmune hemolytic anemia
it is status when in organism development antibodies against own

Слайд 74Autoimmune hemolytic anemia caused by warm-reactive antibodies:
Primary.
Secondary:
1. acute
- viral infections;
- drugs (α-Methyldopa,
Autoimmune hemolytic anemia caused by warm-reactive antibodies:
Primary.
Secondary:
1. acute
- viral infections;
- drugs (α-Methyldopa,

Слайд 75Autoimmune hemolytic anemia caused by cold-reactive antibodies:
Primary.
Secondary:
- mycoplasma infections;
- viral infections;
- lymphoproliferative
Autoimmune hemolytic anemia caused by cold-reactive antibodies:
Primary.
Secondary:
- mycoplasma infections;
- viral infections;
- lymphoproliferative

Слайд 76Autoimmune hemolytic anemia - diagnosis
- Possitive direct Coombs’ test.
Treatment:
Steroids;
Splenectomy;
Immunosupressive agents;
Transfusion.
Autoimmune hemolytic anemia - diagnosis
- Possitive direct Coombs’ test.
Treatment:
Steroids;
Splenectomy;
Immunosupressive agents;
Transfusion.

Слайд 77Hereditary microspherocytosis
1. Pathophysiology:
red cell membrane protein defects (spectrin deficiency) resulting cytoskeleton
Hereditary microspherocytosis
1. Pathophysiology:
red cell membrane protein defects (spectrin deficiency) resulting cytoskeleton

Слайд 78Thalassemia syndromes
a heterogeneous group of inherited anemia characterized by reduced or absent
Thalassemia syndromes
a heterogeneous group of inherited anemia characterized by reduced or absent
Слайд 79Alpha Thalassemia
The alpha thalassemia patient's hemoglobin does not produce enough alpha protein.
Alpha Thalassemia
The alpha thalassemia patient's hemoglobin does not produce enough alpha protein.

Слайд 80One faulty (mutated) gene - there are either no symptoms at all,
One faulty (mutated) gene - there are either no symptoms at all,

Слайд 81Three mutated genes - the patient will have hemoglobin H disease, i.e.
Three mutated genes - the patient will have hemoglobin H disease, i.e.

Слайд 82Mortality/Morbidity
α thalassemia major is a mortal disease, and virtually all affected fetuses
Mortality/Morbidity
α thalassemia major is a mortal disease, and virtually all affected fetuses

Слайд 83Beta Thalassemia
We need two globin genes to make beta globin chains. We
Beta Thalassemia
We need two globin genes to make beta globin chains. We

Слайд 84Severity of beta thalassemia also depends on how many genes are mutated:
If
Severity of beta thalassemia also depends on how many genes are mutated:
If

Слайд 85Signs And Symptoms Of Beta Thalassemia
The majority of infants with beta thalassemia
Signs And Symptoms Of Beta Thalassemia
The majority of infants with beta thalassemia

Слайд 86In patients with various types of β thalassemia, mortality and morbidity vary
In patients with various types of β thalassemia, mortality and morbidity vary

Слайд 87Age
Despite thalassemia's inherited nature, age at onset of symptoms varies significantly. In
Age
Despite thalassemia's inherited nature, age at onset of symptoms varies significantly. In

Слайд 88People with thalassemia mainly have anemia-like symptoms.
Jaundice;
Fatigue;
Pale skin;
Cold hands and feet;
Dyspnea;
Poor
People with thalassemia mainly have anemia-like symptoms.
Jaundice;
Fatigue;
Pale skin;
Cold hands and feet;
Dyspnea;
Poor

Слайд 89Signs And Symptoms Of Alpha Thalassemia
The majority of children with hemoglobin H
Signs And Symptoms Of Alpha Thalassemia
The majority of children with hemoglobin H

Слайд 90Diagnosing of thalassemia
A complete blood count (CBC) - to measure hemoglobin levels,
Diagnosing of thalassemia
A complete blood count (CBC) - to measure hemoglobin levels,

Слайд 91The WBC count is usually elevated in β thalassemia major; this is
The WBC count is usually elevated in β thalassemia major; this is

Слайд 92Treatment of thalassemia
Blood transfusions - this is done to replenish hemoglobin and
Treatment of thalassemia
Blood transfusions - this is done to replenish hemoglobin and

Слайд 93Complications of thalassemia
Iron overload;
Enlarged spleen (splenomegaly);
Infection;
Bone deformities.
Complications of thalassemia
Iron overload;
Enlarged spleen (splenomegaly);
Infection;
Bone deformities.
Слайд 94Congenital sideroblastic anemias
Congenital sideroblastic anemias generally present with lower hemoglobin and more
Congenital sideroblastic anemias
Congenital sideroblastic anemias generally present with lower hemoglobin and more

Слайд 95Etiology
Congenital causes of sideroblastic anemia include the following:
δ-ALAS mutation;
ABC7 mutation;
PSU1 mutation;
Pearson
Etiology
Congenital causes of sideroblastic anemia include the following:
δ-ALAS mutation;
ABC7 mutation;
PSU1 mutation;
Pearson

Слайд 96Major causes of death in cases of sideroblastic anemia are secondary hemochromatosis
Major causes of death in cases of sideroblastic anemia are secondary hemochromatosis

Слайд 97Clinical Presentation
Incoordination (cerebellar symptoms);
Failure of growth;
Diarrhea (malabsorption);
Polyuria, blindness, deafness (associated with DIDMOAD
Clinical Presentation
Incoordination (cerebellar symptoms);
Failure of growth;
Diarrhea (malabsorption);
Polyuria, blindness, deafness (associated with DIDMOAD

Слайд 98Medication history of antibiotics, antituberculous agents, chelators, or chemotherapy;
Ingestion of supplements, especially
Medication history of antibiotics, antituberculous agents, chelators, or chemotherapy;
Ingestion of supplements, especially

Слайд 99Physical Examination
General - Growth retardation in children;
Vital signs – Hypothermia;
Oral - Lead
Physical Examination
General - Growth retardation in children;
Vital signs – Hypothermia;
Oral - Lead

Слайд 100Complete Blood Cell Count and
Peripheral Smear
CBC count usually reveals anemia.
The
Complete Blood Cell Count and
Peripheral Smear
CBC count usually reveals anemia.
The

Слайд 101Sideroblastic anemia also seen in combined vitamin B-12 deficiency with iron deficiency.
Sideroblastic anemia also seen in combined vitamin B-12 deficiency with iron deficiency.

Слайд 102Treatment
Treatment of sideroblastic anemia may include removal of toxic agents;
administration of pyridoxine,
Treatment
Treatment of sideroblastic anemia may include removal of toxic agents;
administration of pyridoxine,

Слайд 103SICKLE CELL ANEMIA
A serious condition in which red blood cells can become
SICKLE CELL ANEMIA
A serious condition in which red blood cells can become
Слайд 104Sickle-shaped cells don’t move easily through blood. They’re stiff and sticky and
Sickle-shaped cells don’t move easily through blood. They’re stiff and sticky and
Слайд 105Normal and Sickled Red Blood Cells
in Blood Vessels
Figure A shows normal
Normal and Sickled Red Blood Cells
in Blood Vessels
Figure A shows normal
Слайд 106People who have sickle cell anemia are born with it; means inherited,
People who have sickle cell anemia are born with it; means inherited,

Слайд 107Inheritance of Sickle Cell Anemia
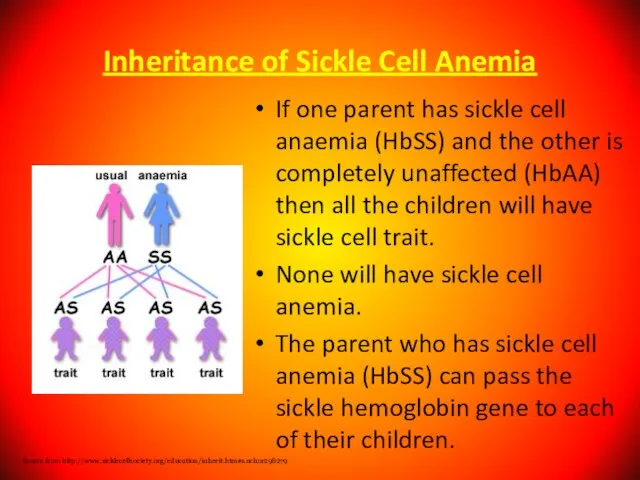
If one parent has sickle cell anaemia (HbSS)
Inheritance of Sickle Cell Anemia
If one parent has sickle cell anaemia (HbSS)
Слайд 108Inheritance of Sickle Cell Anemia
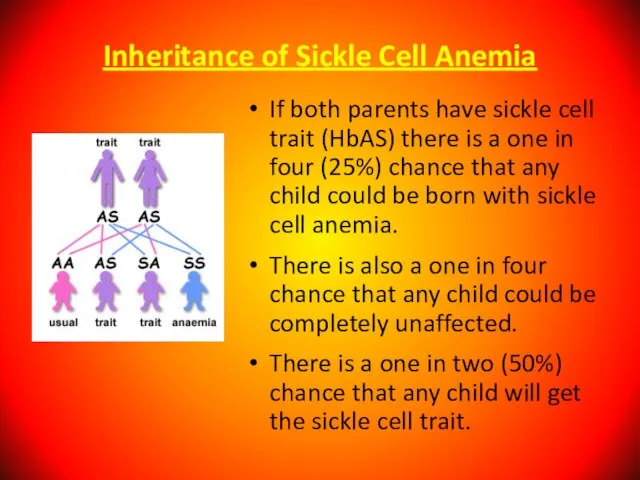
If both parents have sickle cell trait (HbAS)
Inheritance of Sickle Cell Anemia
If both parents have sickle cell trait (HbAS)

Слайд 110Signs and Symptoms
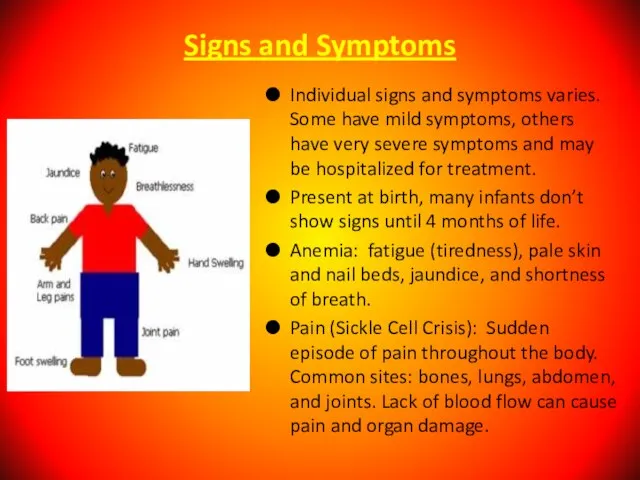
Individual signs and symptoms varies. Some have mild symptoms, others
Signs and Symptoms
Individual signs and symptoms varies. Some have mild symptoms, others
Слайд 111Differential Diagnoses
Acute Anemia;
Carotid Cavernous Fistula;
Hands Rheumatoid Arthritis;
Hemoglobin C Disease;
Hemolytic Anemia;
Legg-Calve-Perthes Disease Imaging;
Leukemias;
Osteomyelitis;
Pulmonary
Differential Diagnoses
Acute Anemia;
Carotid Cavernous Fistula;
Hands Rheumatoid Arthritis;
Hemoglobin C Disease;
Hemolytic Anemia;
Legg-Calve-Perthes Disease Imaging;
Leukemias;
Osteomyelitis;
Pulmonary

Слайд 112Complication of Sickle Cell Anemia
Splenic Crisis
Infections
Acute Chest Syndrome
Delayed growth and puberty in
Complication of Sickle Cell Anemia
Splenic Crisis
Infections
Acute Chest Syndrome
Delayed growth and puberty in

Слайд 113Treatment
Effective treatment is available to help relieve the symptoms and complications of
Treatment
Effective treatment is available to help relieve the symptoms and complications of

Слайд 114Prevention
Identify what can trigger the “Crisis” such as stress, avoid extremes of
Prevention
Identify what can trigger the “Crisis” such as stress, avoid extremes of

Слайд 115Aplastic Anemia
is a syndrome of bone marrow failure characterized by peripheral pancytopenia
Aplastic Anemia
is a syndrome of bone marrow failure characterized by peripheral pancytopenia

Слайд 116Fanconi Anemia
Fanconi anemia is the most frequently reported of the rare inherited
Fanconi Anemia
Fanconi anemia is the most frequently reported of the rare inherited

Слайд 117Multiple congenital anomalies (60-75%):
Short stature,
petechiae and bruises, abnormal skin pigmentation,
Multiple congenital anomalies (60-75%):
Short stature,
petechiae and bruises, abnormal skin pigmentation,

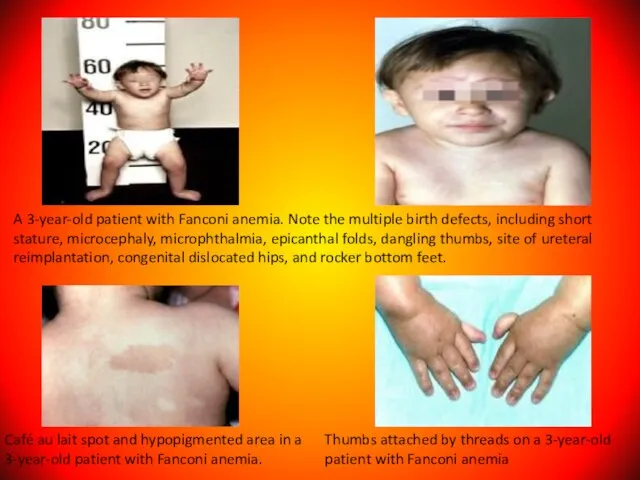
Слайд 118A 3-year-old patient with Fanconi anemia. Note the multiple birth defects, including
A 3-year-old patient with Fanconi anemia. Note the multiple birth defects, including
Слайд 119Bone marrow failure: Thrombocytopenia, leukopenia, or aplastic anemia; most patients with Fanconi
Bone marrow failure: Thrombocytopenia, leukopenia, or aplastic anemia; most patients with Fanconi

Слайд 120Gonads may display the following abnormalities:
Males - Hypogenitalia, undescended testes, hypospadias, abnormal
Gonads may display the following abnormalities:
Males - Hypogenitalia, undescended testes, hypospadias, abnormal

Слайд 121Cancer:
Hematologic malignancies are common with Fanconi anemia and myelodysplasia:
acute myeloid leukemia (AML)
Cancer:
Hematologic malignancies are common with Fanconi anemia and myelodysplasia:
acute myeloid leukemia (AML)

Слайд 122Supportive Care
Treatment is recommended for significant cytopenias, such as hemoglobin less than
Supportive Care
Treatment is recommended for significant cytopenias, such as hemoglobin less than

 Регенерация в сердечно-сосудистой и эндокринной системе
Регенерация в сердечно-сосудистой и эндокринной системе Тренды развития электронной торговли в России
Тренды развития электронной торговли в России Козлова Светлана Георгиевна
Козлова Светлана Георгиевна Лекарственные растения
Лекарственные растения People Trust (Платформа P2P кредитования X5)
People Trust (Платформа P2P кредитования X5) Презентация на тему Афганская война
Презентация на тему Афганская война  Чили
Чили РЕГУЛИРОВАНИЕ КАЧЕСТВА СНАБЖЕНИЯ ЭЛЕКТРОЭНЕРГИЕЙ
РЕГУЛИРОВАНИЕ КАЧЕСТВА СНАБЖЕНИЯ ЭЛЕКТРОЭНЕРГИЕЙ Организация бесплатного и льготного отпуска лекарственных препаратов
Организация бесплатного и льготного отпуска лекарственных препаратов Оптические иллюзии
Оптические иллюзии Трапеция. Площадь трапеции
Трапеция. Площадь трапеции Шерхан Мұртаза
Шерхан Мұртаза ЦЕНТР ПО САПРОПЕЛЮ И IMS инновационные механизированные комплексы очистки водоемов и добычи сапропеля
ЦЕНТР ПО САПРОПЕЛЮ И IMS инновационные механизированные комплексы очистки водоемов и добычи сапропеля Как выбрать шубу?
Как выбрать шубу? Социальный и личностный заказ: возможности согласования образовательных потребностей
Социальный и личностный заказ: возможности согласования образовательных потребностей Художественный образ Гоголя
Художественный образ Гоголя Презентация на тему Путешествие по Италии
Презентация на тему Путешествие по Италии  Теологическая теория происхождения государства и права
Теологическая теория происхождения государства и права Bóg czyni swój lud królestwem Wybór Dawida na króla
Bóg czyni swój lud królestwem Wybór Dawida na króla Физиология кровообращения
Физиология кровообращения Урок алгебры в 10 классе
Урок алгебры в 10 классе Блюда из грибов
Блюда из грибов ООО «ЮниЛекс»
ООО «ЮниЛекс» Сталинград
Сталинград Шлифовальные станки, используемые в производстве фанеры
Шлифовальные станки, используемые в производстве фанеры Дизайн_квартиры_в_доме_Бизнес_класса
Дизайн_квартиры_в_доме_Бизнес_класса Игра "Внимательные глазки"
Игра "Внимательные глазки" Tema_1_5_Myshlenie_i_deyatelnost
Tema_1_5_Myshlenie_i_deyatelnost