- Идиопатические воспалительные миопатии: полимиозит и дерматомиозит

Содержание
- 2. Идиопатические воспалительные миопатии — группа хронических заболеваний, неизвестной этиологии, основным проявлением которых является симметричная мышечная слабость
- 3. Классификация идиопатических воспалительных миопатий (модиф. Miller 1994) 1.Первичный идиопатический полимиозит (ПМ) 2.Первичный идиопатический дерматомиозит (ДМ) 3.Миозит,
- 4. Эпидемиология Распространенность и частота варьирует в различных популяциях. Согласно эпидемиологическим исследованиям, показатели заболеваемости составляют от 2,18
- 5. Дерматомиозит (ДМ) — системное прогрессирующее заболевание с преимущественным поражением поперечно-полосатой и гладкой мускулатуры с нарушением двигательной
- 6. Классификация дерматомиозита (полимиозита) по А. Bohan и Y.Peter (1975) I. Первичный (идиопатический) полимиозит II. Первичный (идиопатический)
- 7. Симптоматика Начало заболевания может быть острым, но чаще симптоматика развивается постепенно, характеризуясь преимущественно кожными и мышечными
- 8. Симптоматика При ПМ поражение кожи отсутствует, но уже с начала заболевания остро или постепенно развивается характерная
- 9. Симптоматика Мышечный синдром проявляется в первую очередь болями в мышцах при движении, прощупывании и даже в
- 10. Симптоматика Возможно течение полимиозита с поражением гладкой мускулатуры глотки, гортани и пищевода. В таких случаях развивается
- 11. Симптоматика Суставной синдром характеризуется поражением лучезапястных суставов и мелких суставов кисти. Реже при полимиозите наблюдается поражение
- 12. Клинические признаки Повышение температуры тела Поражение кожи: эритема периорбитальный отек капилляриты отек Синдром Рейно Генерализованное поражение
- 13. Диагностические критерии ПМ/ДМ (Bohan, Peter 1975) 1. Симметричная проксимальная слабость мышц плечевого и тазового пояса, нарастающая
- 14. Лабораторно- инструментальные методы исследования Увеличение КФК, АЛТ, АСТ, ЛДГ. Аутоантитела обнаруживаются в сыворотке пациентов 50% ПМ/ДМ.
- 15. Миозит-специфические антитела выявляются только при ИВМ и далее маркеруют их клинические фенотипы. К ним относятся анти-Мi-2,
- 16. Морфологическое исследование. Дерматомиозит является комплемент-зависимой микроангиопатией, ведущей к разрушению капилляров, повышенной инфильтрации плазмой и воспалительными клетками
- 17. При полимиозите наблюдаются множественные очаги воспаления, где выявляются CD8+Т-клетки, которые проникают в неизмененные мышечные волокна, экспрессирующие
- 18. Алгоритм диагностического поиска и дифференциальный диагноз у пациентов с мышечной слабостью (не получающих ГК):
- 20. Методы оценки мышечной силы. Мануальное тестирование силы проксимальных и аксиальных мышц проводится согласно рекомендациям IMACS и
- 21. Лечение ПМ/ДМ !!! У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых, затрагивая мышечную ткань, может
- 22. Лечение ПМ/ДМ • Раннее начало терапии (в течение первых 3-х месяцев от начала симптомов) ассоциируется с
- 23. Лечение ПМ/ДМ • Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца. • Снижение дозы ГК
- 24. Лечение ПМ/ДМ • Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ и не служит
- 25. Лечение ПМ/ДМ Потенциальные показания к подключению иммуносупрессивной терапии : • Принадлежность больных к клинико-иммунологическим подтипам ПМ/ДМ,
- 26. Лечение ПМ/ДМ • Метотрексат по 7,5–25 мг/нед внутрь или внутривенно (при недостаточной эффективности или плохой переносимости
- 27. Лечение ПМ/ДМ Плазмаферез следует использовать главным образом у больных с тяжёлым, резистентным к другим метода лечения
- 28. Ведение пациентов ПМ/ДМ с хроническим течением болезни, связанным с неадекватно малой инициальной дозой ГК
- 30. Скачать презентацию
Слайд 2Идиопатические воспалительные миопатии — группа хронических заболеваний, неизвестной этиологии, основным проявлением которых
Идиопатические воспалительные миопатии — группа хронических заболеваний, неизвестной этиологии, основным проявлением которых

Слайд 3Классификация идиопатических воспалительных миопатий (модиф. Miller 1994)
1.Первичный идиопатический полимиозит (ПМ)
2.Первичный идиопатический дерматомиозит
Классификация идиопатических воспалительных миопатий (модиф. Miller 1994)
1.Первичный идиопатический полимиозит (ПМ)
2.Первичный идиопатический дерматомиозит

Слайд 4Эпидемиология
Распространенность и частота варьирует в различных популяциях. Согласно эпидемиологическим исследованиям, показатели заболеваемости
Эпидемиология
Распространенность и частота варьирует в различных популяциях. Согласно эпидемиологическим исследованиям, показатели заболеваемости

Слайд 5Дерматомиозит (ДМ) — системное прогрессирующее заболевание с преимущественным поражением поперечно-полосатой и гладкой
Дерматомиозит (ДМ) — системное прогрессирующее заболевание с преимущественным поражением поперечно-полосатой и гладкой

Слайд 6Классификация дерматомиозита (полимиозита) по А. Bohan и Y.Peter (1975) I. Первичный (идиопатический)
Классификация дерматомиозита (полимиозита) по А. Bohan и Y.Peter (1975) I. Первичный (идиопатический)

Слайд 7Симптоматика
Начало заболевания может быть острым, но чаще симптоматика развивается постепенно, характеризуясь преимущественно
Симптоматика
Начало заболевания может быть острым, но чаще симптоматика развивается постепенно, характеризуясь преимущественно

Слайд 8Симптоматика
При ПМ поражение кожи отсутствует, но уже с начала заболевания остро или
Симптоматика
При ПМ поражение кожи отсутствует, но уже с начала заболевания остро или

Слайд 9Симптоматика
Мышечный синдром проявляется в первую очередь болями в мышцах при движении, прощупывании
Симптоматика
Мышечный синдром проявляется в первую очередь болями в мышцах при движении, прощупывании

Слайд 10Симптоматика
Возможно течение полимиозита с поражением гладкой мускулатуры глотки, гортани и пищевода. В
Симптоматика
Возможно течение полимиозита с поражением гладкой мускулатуры глотки, гортани и пищевода. В

Слайд 11Симптоматика
Суставной синдром характеризуется поражением лучезапястных суставов и мелких суставов кисти. Реже при
Симптоматика
Суставной синдром характеризуется поражением лучезапястных суставов и мелких суставов кисти. Реже при

Слайд 12Клинические признаки
Повышение температуры тела
Поражение кожи:
эритема
периорбитальный отек
капилляриты
отек
Синдром Рейно
Генерализованное поражение
Клинические признаки
Повышение температуры тела
Поражение кожи:
эритема
периорбитальный отек
капилляриты
отек
Синдром Рейно
Генерализованное поражение

Слайд 13Диагностические критерии ПМ/ДМ (Bohan, Peter 1975)
1. Симметричная проксимальная слабость мышц плечевого и
Диагностические критерии ПМ/ДМ (Bohan, Peter 1975)
1. Симметричная проксимальная слабость мышц плечевого и

Слайд 14Лабораторно- инструментальные методы исследования
Увеличение КФК, АЛТ, АСТ, ЛДГ.
Аутоантитела обнаруживаются в сыворотке
Лабораторно- инструментальные методы исследования
Увеличение КФК, АЛТ, АСТ, ЛДГ.
Аутоантитела обнаруживаются в сыворотке

Слайд 15Миозит-специфические антитела выявляются только при ИВМ и далее маркеруют их клинические фенотипы.
Миозит-специфические антитела выявляются только при ИВМ и далее маркеруют их клинические фенотипы.

Слайд 16Морфологическое исследование.
Дерматомиозит является комплемент-зависимой микроангиопатией, ведущей к разрушению капилляров, повышенной инфильтрации
Морфологическое исследование.
Дерматомиозит является комплемент-зависимой микроангиопатией, ведущей к разрушению капилляров, повышенной инфильтрации

Слайд 17При полимиозите наблюдаются множественные очаги воспаления, где выявляются CD8+Т-клетки, которые проникают в
При полимиозите наблюдаются множественные очаги воспаления, где выявляются CD8+Т-клетки, которые проникают в

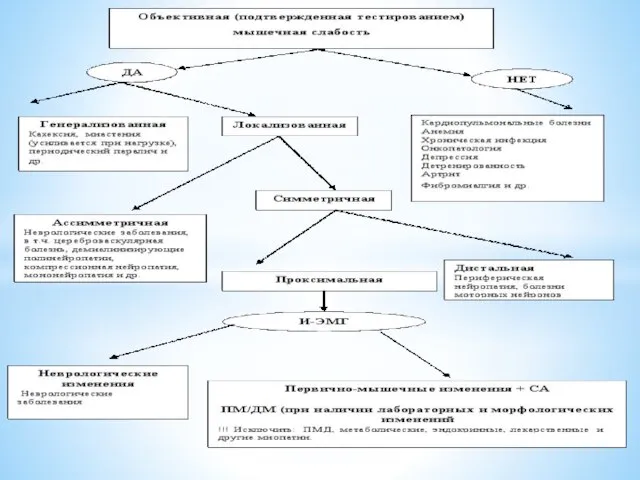
Слайд 18Алгоритм диагностического поиска и дифференциальный диагноз у пациентов с мышечной слабостью (не
Алгоритм диагностического поиска и дифференциальный диагноз у пациентов с мышечной слабостью (не

Слайд 20 Методы оценки мышечной силы.
Мануальное тестирование силы проксимальных и аксиальных мышц проводится
Методы оценки мышечной силы.
Мануальное тестирование силы проксимальных и аксиальных мышц проводится

Слайд 21 Лечение ПМ/ДМ
!!! У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых, затрагивая
Лечение ПМ/ДМ
!!! У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых, затрагивая

Слайд 22 Лечение ПМ/ДМ
• Раннее начало терапии (в течение первых 3-х месяцев от начала симптомов)
Лечение ПМ/ДМ
• Раннее начало терапии (в течение первых 3-х месяцев от начала симптомов)

Слайд 23 Лечение ПМ/ДМ
• Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца.
• Снижение дозы ГК
Лечение ПМ/ДМ
• Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца.
• Снижение дозы ГК

Слайд 24 Лечение ПМ/ДМ
• Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ и
Лечение ПМ/ДМ
• Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ и

Слайд 25 Лечение ПМ/ДМ
Потенциальные показания к подключению иммуносупрессивной терапии :
• Принадлежность больных к клинико-иммунологическим подтипам
Лечение ПМ/ДМ
Потенциальные показания к подключению иммуносупрессивной терапии :
• Принадлежность больных к клинико-иммунологическим подтипам

Слайд 26 Лечение ПМ/ДМ
• Метотрексат по 7,5–25 мг/нед внутрь или внутривенно (при недостаточной эффективности или
Лечение ПМ/ДМ
• Метотрексат по 7,5–25 мг/нед внутрь или внутривенно (при недостаточной эффективности или

Слайд 27 Лечение ПМ/ДМ
Плазмаферез следует использовать главным образом у больных с тяжёлым, резистентным к
Лечение ПМ/ДМ
Плазмаферез следует использовать главным образом у больных с тяжёлым, резистентным к

Слайд 28 Ведение пациентов ПМ/ДМ с хроническим течением болезни, связанным с неадекватно малой инициальной
Ведение пациентов ПМ/ДМ с хроническим течением болезни, связанным с неадекватно малой инициальной

 Что такое мультимедиа 8 класс
Что такое мультимедиа 8 класс Презентация на тему Влияние шума и музыки на здоровье человека
Презентация на тему Влияние шума и музыки на здоровье человека Мультфильм Умная собачка Соня
Мультфильм Умная собачка Соня ПРОГРАММА РАЗВИТИЯ ГОУ СОШ №16 на 2011-2015 гг. «КАЧЕСТВО ОБРАЗОВАНИЯ ЧЕРЕЗ МЕТАМЕТОДИКУ»
ПРОГРАММА РАЗВИТИЯ ГОУ СОШ №16 на 2011-2015 гг. «КАЧЕСТВО ОБРАЗОВАНИЯ ЧЕРЕЗ МЕТАМЕТОДИКУ» Презентация на тему лягушка-путешественница 3 класс
Презентация на тему лягушка-путешественница 3 класс Информационные технологии в жилищно-коммунальном хозяйстве Финляндии
Информационные технологии в жилищно-коммунальном хозяйстве Финляндии Презентация на тему У каждого ребенка свой дар
Презентация на тему У каждого ребенка свой дар Этап: экономическая теория, анализ, практика
Этап: экономическая теория, анализ, практика Турбокомпрессор ЯМЗ (ТКР ЯМЗ KG-90). ООО Силовые агрегаты – группа ГАЗ
Турбокомпрессор ЯМЗ (ТКР ЯМЗ KG-90). ООО Силовые агрегаты – группа ГАЗ Презентация на тему Анализ опасности поражения электрическим током
Презентация на тему Анализ опасности поражения электрическим током  Рынок взыскания задолженности и законопроект «О деятельности по взысканию просроченной задолженности»
Рынок взыскания задолженности и законопроект «О деятельности по взысканию просроченной задолженности» Дружная семья
Дружная семья Презентация на тему Сосновый лес
Презентация на тему Сосновый лес  ФГБОУ ВО Кемеровский государственный институт культуры. Отчет по учебной практике
ФГБОУ ВО Кемеровский государственный институт культуры. Отчет по учебной практике Где ума набраться…
Где ума набраться… Моя индивидуальность
Моя индивидуальность Общая характеристика оральных гормональных контрацептивов
Общая характеристика оральных гормональных контрацептивов Зимние Олимпийские игры
Зимние Олимпийские игры Функции речи
Функции речи Шпа Музыкальная интуиция
Шпа Музыкальная интуиция Оценка достижений
Оценка достижений Великие левши
Великие левши Виват, Россия! Городской конкурс военно-патриотической песни
Виват, Россия! Городской конкурс военно-патриотической песни 1
1 Концепция Винного Фестиваля
Концепция Винного Фестиваля Порядок осуществления закупок Предприятием ФГУП Калужское
Порядок осуществления закупок Предприятием ФГУП Калужское Петербург Раскольникова
Петербург Раскольникова Презентация на тему Наши статусы, или в какие группы общества мы входим (7 класс)
Презентация на тему Наши статусы, или в какие группы общества мы входим (7 класс)