- Нервно-мышечные заболевания

Содержание
- 2. Нервно-мышечные заболевания большая гетерогенная группа наследственных и ненаследственных заболеваний, характеризующихся нарушением функций: а)мышечной системы – миопатии
- 4. Эпидемиология Суммарная распространенность нервно-мышечных заболеваний (НМЗ) составляет 27,2 на 100 000 человек; при этом «ядро» нозологического
- 5. Первичные наследственные миопатии- прогрессирующие мышечные дистрофии: это болезни , при которых расстройства метаболизма ведут к прогрессирующей
- 6. Классификация: псевдогипертрофическая Дюшенна; псевдогипертрофическая Беккера; Эмери-Дрейфуса- Хогана; Роттауфа- Мортье –Бейра (фиброзирующая миопатия); ювениальная Эрба-Рота; окулярная (хроническая
- 7. Этиология. У больных ПМД выявляется врожденный структурный дефект мышечной ткани: при ПМД Дюшенна- дефект гена, отвечающего
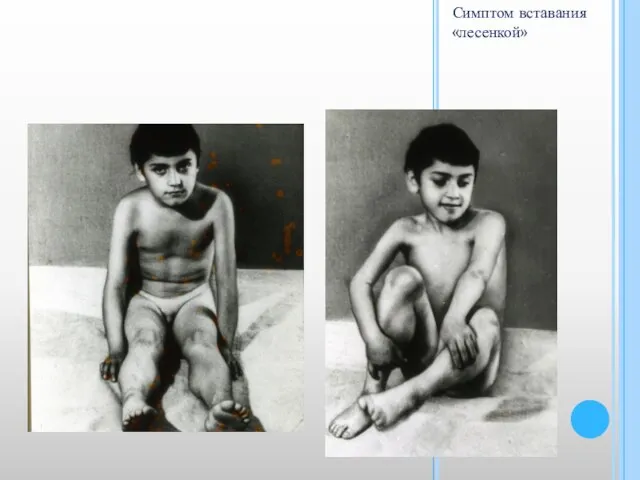
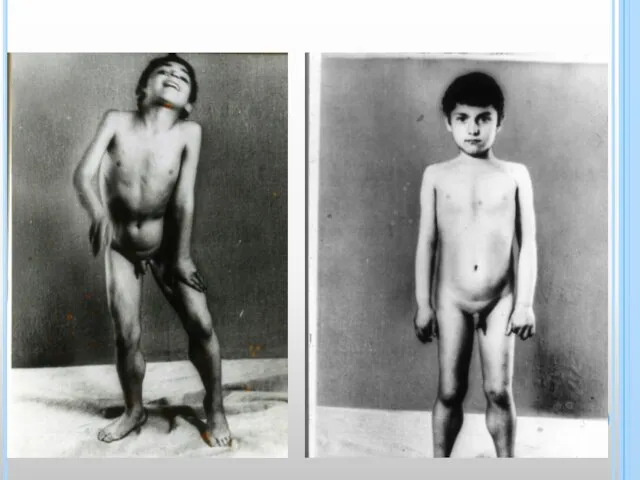
- 8. КЛИНИКА «Утиная походка» – пациент при ходьбе «переваливается» с ноги на ногу из-за слабости ягодичных мышц;
- 9. КЛИНИКА «лягушачий живот»- низкий тонус и гипотрофия мышц живота ; лицо «сфинкса»- слабость и гипотрофия мыщц
- 10. Симптом вставания «лесенкой»
- 14. «крыловидные лопатки» «кифосколиоз»
- 15. Метаболические миопатии ММ представляют собой обширную группу наследственных и приобретенных заболеваний, связанных с нарушением обмена веществ
- 16. ПРИЧИНЫ Наследственная предрасположенность (аутосомно-рецессивно) эндокринные заболевания хронические интоксикации хронические болезни почек заболевания печени синдром мальабсорбции Врожденных
- 17. Классификация: Гликогенозы ( дефекты метаболизма гликогена) Липидные ( дефекты метаболизма липидов) Митохондриальные ( дефекты митохондриального окислительного
- 18. Классификация наследственных форм метаболический миопатий Нарушения метаболизма мышечного гликогена Дефицит кислой мальтазы (болезнь Помпе) Дефицит ветвящего
- 19. Недостаток кислой мальтазы в лизосомах Болезнь Помпе ( гликогеноз 2 типа)- относится к редким мультисистемным наследственным
- 20. Болезнь Помпе Первые признаки и симптомы МБП появляются уже на 2-3 месяцах жизни. При осмотре на
- 21. Недостаток миофосфорилазы. Миофосфорилаза (гликогенфосфорилаза) расщепляет гликоген до глюкозо-1-фосфата и таким образом участвует в выработке энергии для
- 22. Митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды( MELAS- синдром). В основе патогенеза- точковые мутации митохондриальных ДНК, контролирующих дыхательную
- 23. Клинические симптомы (1994 г): Непереносимость физических нагрузок(100%) Дебют заболевания в возрасте до 40 лет (100%) Инсультоподобные
- 24. Клинические симптомы (1994 г): Другие симптомы: Кальцификация базальных ганглиев (45%) Отягощенный семейный анамнез(44%) Миоклонус(38%) Мозжечковые симптомы
- 25. Лабораторные и функциональные исследования: Лактат-ацидоз в крови и ЦСЖ ( у половины больных повышено содержание лактат
- 26. Лабораторные и функциональные исследования: ЭКГ : 12% больных – синдром Вольфа-Паркинсона – Уайта КТ-мозга ( зоны
- 27. Миотонии сопровождаются нарушением расслабления мышц после их сокращения при целенаправленных движениях. Классификация: миотония Томсена; миотония Беккера;
- 28. Клиника симптом мышечного валика- при ударе молоточком по мышце на месте удара некоторое время сохраняется ямка
- 29. Миастении характеризуются нарушением нервно-мышечной передачи и проявляются патологической мышечной утомляемостью, слабостью.
- 30. Этиология и патогенез
- 31. Классификация Неонатальная миастения ; Ювениальная ( врожденная , ранняя детская, юношеская); Миастения взрослых .
- 32. Формы миастении взрослых: глазная скелетно-мышечная молниеносная глоточно-лицевая краниальная
- 35. Спинальная амиотрофия -хар-ся дегенерацией двигательных нейронов передних рогов спинного мозга. Типы: I типа (болезнь Верднига-Гоффмана); II
- 36. Электромиограмма при спинальной амиотрофии — «ритм частокола» Амиотрофия III типа
- 37. Невральная амиотрофия Шарко-Мари-Тута преобладают атрофические изменения мышц дистальных отделов конечностей ; страдают разгибатели голени, а также
- 38. Диагностика биохимические; электрофизиологические; патоморфологические; ДНК-диагностика .
- 40. Скачать презентацию
Слайд 2Нервно-мышечные заболевания
большая гетерогенная группа наследственных и ненаследственных заболеваний, характеризующихся нарушением функций:
а)мышечной
Нервно-мышечные заболевания
большая гетерогенная группа наследственных и ненаследственных заболеваний, характеризующихся нарушением функций: а)мышечной


Слайд 4Эпидемиология
Суммарная распространенность нервно-мышечных заболеваний (НМЗ) составляет 27,2 на 100 000 человек;
при этом «ядро» нозологического спектра
Эпидемиология
Суммарная распространенность нервно-мышечных заболеваний (НМЗ) составляет 27,2 на 100 000 человек;
при этом «ядро» нозологического спектра

Слайд 5Первичные наследственные миопатии- прогрессирующие мышечные дистрофии:
это болезни , при которых расстройства метаболизма
Первичные наследственные миопатии- прогрессирующие мышечные дистрофии:
это болезни , при которых расстройства метаболизма

Слайд 6 Классификация:
псевдогипертрофическая Дюшенна;
псевдогипертрофическая Беккера;
Эмери-Дрейфуса- Хогана;
Роттауфа- Мортье –Бейра (фиброзирующая миопатия);
ювениальная Эрба-Рота;
окулярная (хроническая прогрессирующая
Классификация:
псевдогипертрофическая Дюшенна;
псевдогипертрофическая Беккера;
Эмери-Дрейфуса- Хогана;
Роттауфа- Мортье –Бейра (фиброзирующая миопатия);
ювениальная Эрба-Рота;
окулярная (хроническая прогрессирующая

Слайд 7 Этиология.
У больных ПМД выявляется врожденный структурный дефект мышечной ткани:
при ПМД
Этиология.
У больных ПМД выявляется врожденный структурный дефект мышечной ткани:
при ПМД

Слайд 8КЛИНИКА
«Утиная походка» – пациент при ходьбе «переваливается» с ноги на ногу из-за
КЛИНИКА
«Утиная походка» – пациент при ходьбе «переваливается» с ноги на ногу из-за

Слайд 9 КЛИНИКА
«лягушачий живот»- низкий тонус и гипотрофия мышц живота ;
лицо «сфинкса»- слабость
КЛИНИКА
«лягушачий живот»- низкий тонус и гипотрофия мышц живота ;
лицо «сфинкса»- слабость
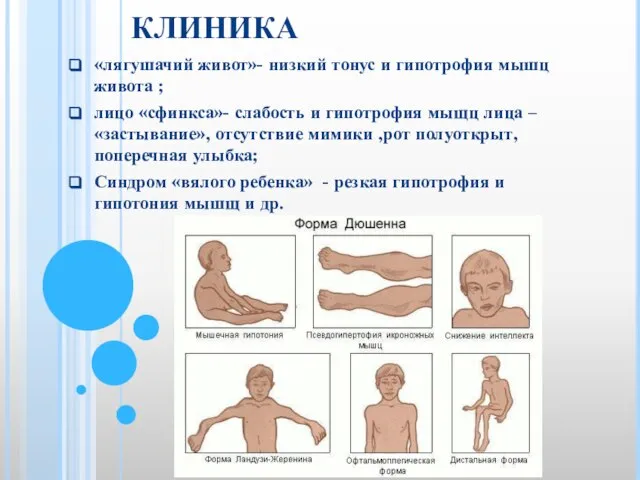
Слайд 10Симптом вставания «лесенкой»
Симптом вставания «лесенкой»


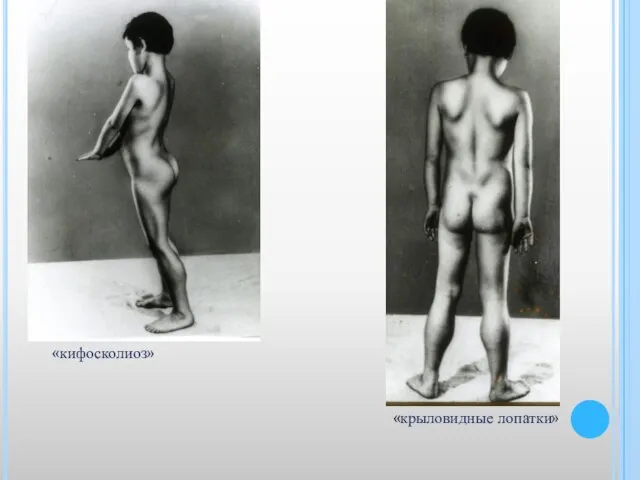
Слайд 14«крыловидные лопатки»
«кифосколиоз»
«крыловидные лопатки»
«кифосколиоз»
Слайд 15Метаболические миопатии
ММ представляют собой обширную группу наследственных и приобретенных заболеваний, связанных
Метаболические миопатии
ММ представляют собой обширную группу наследственных и приобретенных заболеваний, связанных

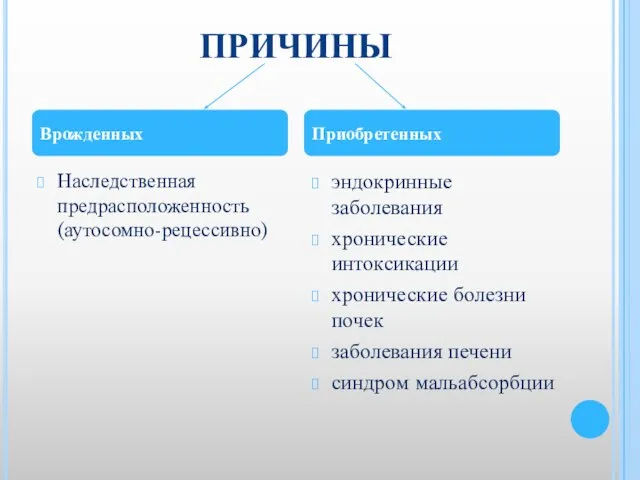
Слайд 16ПРИЧИНЫ
Наследственная предрасположенность (аутосомно-рецессивно)
эндокринные заболевания
хронические интоксикации
хронические болезни почек
заболевания печени
синдром мальабсорбции
Врожденных
Приобретенных
ПРИЧИНЫ
Наследственная предрасположенность (аутосомно-рецессивно)
эндокринные заболевания
хронические интоксикации
хронические болезни почек
заболевания печени
синдром мальабсорбции
Врожденных
Приобретенных
Слайд 17Классификация:
Гликогенозы ( дефекты метаболизма гликогена)
Липидные ( дефекты метаболизма липидов)
Митохондриальные ( дефекты
Классификация:
Гликогенозы ( дефекты метаболизма гликогена)
Липидные ( дефекты метаболизма липидов)
Митохондриальные ( дефекты

Слайд 18Классификация наследственных форм метаболический миопатий
Нарушения метаболизма мышечного гликогена
Дефицит кислой мальтазы (болезнь Помпе)
Дефицит
Классификация наследственных форм метаболический миопатий
Нарушения метаболизма мышечного гликогена
Дефицит кислой мальтазы (болезнь Помпе)
Дефицит

Слайд 19Недостаток кислой мальтазы в лизосомах
Болезнь Помпе ( гликогеноз 2 типа)- относится к
Недостаток кислой мальтазы в лизосомах
Болезнь Помпе ( гликогеноз 2 типа)- относится к

Слайд 20Болезнь Помпе
Первые признаки и симптомы МБП появляются уже на 2-3 месяцах жизни.
Болезнь Помпе
Первые признаки и симптомы МБП появляются уже на 2-3 месяцах жизни.

Слайд 21Недостаток миофосфорилазы.
Миофосфорилаза (гликогенфосфорилаза) расщепляет гликоген до глюкозо-1-фосфата и таким образом участвует в
Недостаток миофосфорилазы.
Миофосфорилаза (гликогенфосфорилаза) расщепляет гликоген до глюкозо-1-фосфата и таким образом участвует в

Слайд 22Митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды( MELAS- синдром).
В основе патогенеза- точковые мутации митохондриальных
Митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды( MELAS- синдром).
В основе патогенеза- точковые мутации митохондриальных

Слайд 23Клинические симптомы (1994 г):
Непереносимость физических нагрузок(100%)
Дебют заболевания в возрасте до 40 лет
Клинические симптомы (1994 г):
Непереносимость физических нагрузок(100%)
Дебют заболевания в возрасте до 40 лет

Слайд 24Клинические симптомы (1994 г):
Другие симптомы:
Кальцификация базальных ганглиев (45%)
Отягощенный семейный анамнез(44%)
Миоклонус(38%)
Мозжечковые симптомы (33%)
Эпизоды
Клинические симптомы (1994 г):
Другие симптомы:
Кальцификация базальных ганглиев (45%)
Отягощенный семейный анамнез(44%)
Миоклонус(38%)
Мозжечковые симптомы (33%)
Эпизоды

Слайд 25Лабораторные и функциональные исследования:
Лактат-ацидоз в крови и ЦСЖ ( у половины больных
Лабораторные и функциональные исследования:
Лактат-ацидоз в крови и ЦСЖ ( у половины больных

Слайд 26Лабораторные и функциональные исследования:
ЭКГ : 12% больных – синдром Вольфа-Паркинсона – Уайта
КТ-мозга
Лабораторные и функциональные исследования:
ЭКГ : 12% больных – синдром Вольфа-Паркинсона – Уайта
КТ-мозга

Слайд 27Миотонии
сопровождаются нарушением расслабления мышц после их сокращения при целенаправленных движениях.
Классификация:
миотония Томсена;
миотония Беккера;
Дистрофическая
Миотонии
сопровождаются нарушением расслабления мышц после их сокращения при целенаправленных движениях.
Классификация:
миотония Томсена;
миотония Беккера;
Дистрофическая

Слайд 28Клиника
симптом мышечного валика- при ударе молоточком по мышце на месте удара некоторое
Клиника
симптом мышечного валика- при ударе молоточком по мышце на месте удара некоторое

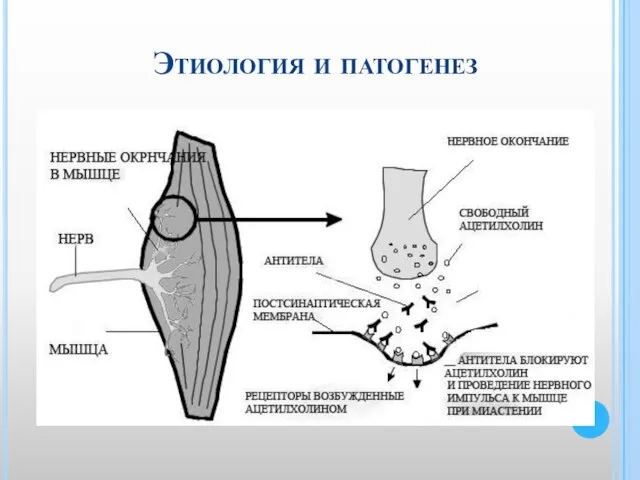
Слайд 29Миастении
характеризуются нарушением нервно-мышечной передачи и проявляются патологической мышечной утомляемостью, слабостью.
Миастении
характеризуются нарушением нервно-мышечной передачи и проявляются патологической мышечной утомляемостью, слабостью.

Слайд 30Этиология и патогенез
Этиология и патогенез
Слайд 31Классификация
Неонатальная миастения ;
Ювениальная ( врожденная , ранняя детская, юношеская);
Миастения взрослых .
Классификация
Неонатальная миастения ;
Ювениальная ( врожденная , ранняя детская, юношеская);
Миастения взрослых .

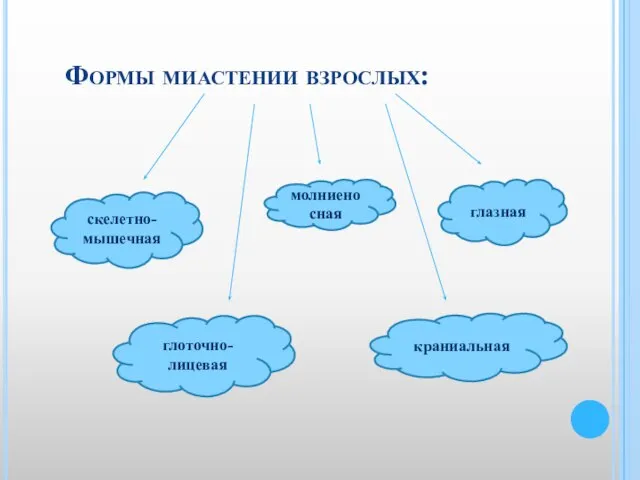
Слайд 32Формы миастении взрослых:
глазная
скелетно-мышечная
молниеносная
глоточно-лицевая
краниальная
Формы миастении взрослых:
глазная
скелетно-мышечная
молниеносная
глоточно-лицевая
краниальная


Слайд 35Спинальная амиотрофия
-хар-ся дегенерацией двигательных нейронов передних рогов спинного мозга.
Типы:
I типа (болезнь Верднига-Гоффмана);
II
Спинальная амиотрофия
-хар-ся дегенерацией двигательных нейронов передних рогов спинного мозга.
Типы:
I типа (болезнь Верднига-Гоффмана);
II

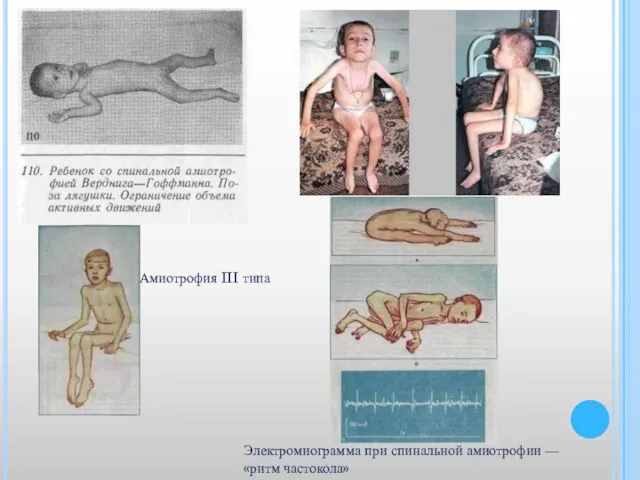
Слайд 36Электромиограмма при спинальной амиотрофии — «ритм частокола»
Амиотрофия III типа
Электромиограмма при спинальной амиотрофии — «ритм частокола»
Амиотрофия III типа
Слайд 37Невральная амиотрофия Шарко-Мари-Тута
преобладают атрофические изменения мышц дистальных отделов конечностей ;
страдают разгибатели голени,
Невральная амиотрофия Шарко-Мари-Тута
преобладают атрофические изменения мышц дистальных отделов конечностей ;
страдают разгибатели голени,

Слайд 38Диагностика
биохимические;
электрофизиологические;
патоморфологические;
ДНК-диагностика .
Диагностика
биохимические;
электрофизиологические;
патоморфологические;
ДНК-диагностика .

 Проблемы урегулирования межорганизационных конфликтов в предпринимательских сетях
Проблемы урегулирования межорганизационных конфликтов в предпринимательских сетях Физиология автономной нервной системы
Физиология автономной нервной системы Презентация на тему Викторина ПДД
Презентация на тему Викторина ПДД  Саратовское региональное отделение Российского благотворительного фонда «Нет алкоголизму и наркомании» Учебный центр по снижен
Саратовское региональное отделение Российского благотворительного фонда «Нет алкоголизму и наркомании» Учебный центр по снижен Световой будильник-жалюзи
Световой будильник-жалюзи Презентация на тему Экологические проблемы России
Презентация на тему Экологические проблемы России  Словообразование и орфография
Словообразование и орфография Хаос
Хаос Профессия Учитель!!! Галина Васильевна – наш классный руководитель.
Профессия Учитель!!! Галина Васильевна – наш классный руководитель. Бихевиоризм в теории управления
Бихевиоризм в теории управления Расширенное Участие Trust Fund Кодексав деятельности Кодекса
Расширенное Участие Trust Fund Кодексав деятельности Кодекса Русско-турецкая война 1877-1878 (8 класс)
Русско-турецкая война 1877-1878 (8 класс) Как психологу повлиять на трудного клиента
Как психологу повлиять на трудного клиента Развитие стран Западной Европы и США Во второй половине XX века
Развитие стран Западной Европы и США Во второй половине XX века Остров Кука
Остров Кука Итоги осуществления закупок товаров, работ и услуг для обеспечения государственных и муниципальных нужд города Чебоксары
Итоги осуществления закупок товаров, работ и услуг для обеспечения государственных и муниципальных нужд города Чебоксары Феномен науки и законы её развития. Педагогика как наука. Лекция 3
Феномен науки и законы её развития. Педагогика как наука. Лекция 3 1-_kultur_ve_destinasyon
1-_kultur_ve_destinasyon Фольклорный коллектив Ӧмидз тусьяс (Ягодки – малинки) с. Усть - Лыжа
Фольклорный коллектив Ӧмидз тусьяс (Ягодки – малинки) с. Усть - Лыжа Работы МДК. Шрифты
Работы МДК. Шрифты Завоевание Обетованной земли
Завоевание Обетованной земли Виды собственности
Виды собственности Парные согласные на конце слов
Парные согласные на конце слов Симметрия “бянь-变” и хуа-花” в орнаменталистике Древнего Китая
Симметрия “бянь-变” и хуа-花” в орнаменталистике Древнего Китая Проектирование волоконно-оптической системы передачи на участке г. Тобольск - с. Ярково
Проектирование волоконно-оптической системы передачи на участке г. Тобольск - с. Ярково Конституция Российской Федерации. Конкурс Молодец
Конституция Российской Федерации. Конкурс Молодец 5 Финансовая система
5 Финансовая система Центральная библиотека Пущинского научного центра РАНВасильчиков Виктор Всеволодович
Центральная библиотека Пущинского научного центра РАНВасильчиков Виктор Всеволодович