- Лейкодистрофии

Содержание
- 2. Определение Лейкодистрофия - (греч. leukos белый + дистрофия) — группа наследственных заболеваний, характеризующихся прогрессирующей дистрофией белого
- 3. Классификация лейкодистрофий Адренолейкодистрофия Метахроматическая лейкодистрофия Глобоидноклеточная лейкодистрофия (болезнь Краббе) Болезнь Канаван Болезнь Пелицеуса-Мерцбахера Болезнь Александера
- 4. Адренолейкодистрофия Нарушение бета-окисления жирных кислот в пероксисомах, которое ведет к накоплению длинноцепочечных ЖК в тканях всего
- 5. МРТ
- 6. Клиническая картина и диагностика АЛД Клиническая картина при АЛД сложна для диагностики, так как данная патология
- 7. Лечение АЛД Диетотерапия: ограничение потребления продуктов, содержащих насыщенные ЖК, прием препаратов ненасыщенных ЖК (масло Лоренцо). Трансплантация
- 8. Метахроматическая лейкодистрофия МЛД развивается из-за недостаточности арилсульфатазы А2. Без этого фермента сульфатиды накапливаются во многих тканях
- 9. Диагностика МЛД Сульфатиды обнаруживают в нервных клетках, сетчатке глаза, шванновской оболочке нервных волокон, в канальцах почек.
- 10. Глобоидноклеточная лейкодистрофия (болезнь Краббе) Редкое нарушение, которое поражает миелиновую оболочку нерва и наследуется по аутосомно-рецессивному типу.
- 11. Болезнь Пелицеуса-Мерцбахера Характеризуется формированием островков интактного миелина в областях с тяжелой демиелинизацией ("леопардов кожа"). Болезнь начинается
- 12. Болезнь Канаван Наследуется по аутосомно-рецессивному типу. Поврежденный ген находится в 17 хромосоме и кодирует фермент аспартоацилазу.
- 13. Мозг четырехлетнего мальчика с болезнью Александера. Видна перивентрикулярная демиелинизация (коричневое обесцвечивание вокруг желудочков).
- 14. Болезнь Александера Развивается в связи с мутацией в гене, который кодирует глиальный фибриллярный кислотный протеин и
- 16. Скачать презентацию
Слайд 2Определение
Лейкодистрофия - (греч. leukos белый + дистрофия) — группа наследственных заболеваний, характеризующихся
Определение
Лейкодистрофия - (греч. leukos белый + дистрофия) — группа наследственных заболеваний, характеризующихся

Слайд 3Классификация лейкодистрофий
Адренолейкодистрофия
Метахроматическая лейкодистрофия
Глобоидноклеточная лейкодистрофия (болезнь Краббе)
Болезнь Канаван
Болезнь Пелицеуса-Мерцбахера
Болезнь Александера
Классификация лейкодистрофий
Адренолейкодистрофия
Метахроматическая лейкодистрофия
Глобоидноклеточная лейкодистрофия (болезнь Краббе)
Болезнь Канаван
Болезнь Пелицеуса-Мерцбахера
Болезнь Александера

Слайд 4Адренолейкодистрофия
Нарушение бета-окисления жирных кислот в пероксисомах, которое ведет к накоплению длинноцепочечных ЖК
Адренолейкодистрофия
Нарушение бета-окисления жирных кислот в пероксисомах, которое ведет к накоплению длинноцепочечных ЖК

Слайд 5МРТ
МРТ
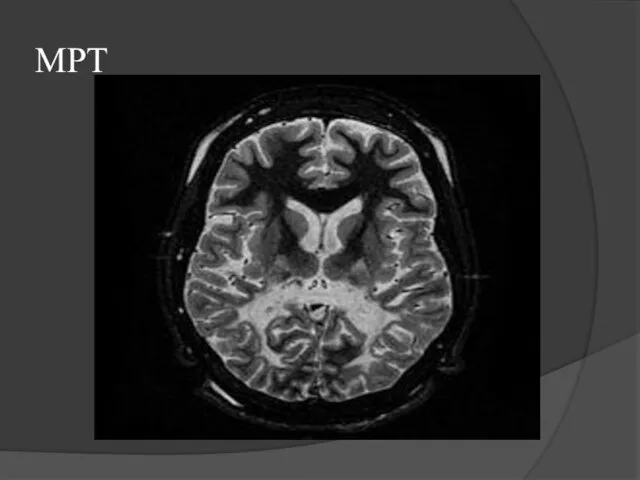
Слайд 6Клиническая картина и диагностика АЛД
Клиническая картина при АЛД сложна для диагностики, так
Клиническая картина и диагностика АЛД
Клиническая картина при АЛД сложна для диагностики, так

Слайд 7Лечение АЛД
Диетотерапия: ограничение потребления продуктов, содержащих насыщенные ЖК, прием препаратов ненасыщенных ЖК
Лечение АЛД
Диетотерапия: ограничение потребления продуктов, содержащих насыщенные ЖК, прием препаратов ненасыщенных ЖК

Слайд 8Метахроматическая лейкодистрофия
МЛД развивается из-за недостаточности арилсульфатазы А2. Без этого фермента сульфатиды накапливаются
Метахроматическая лейкодистрофия
МЛД развивается из-за недостаточности арилсульфатазы А2. Без этого фермента сульфатиды накапливаются

Слайд 9Диагностика МЛД
Сульфатиды обнаруживают в нервных клетках, сетчатке глаза, шванновской оболочке нервных волокон,
Диагностика МЛД
Сульфатиды обнаруживают в нервных клетках, сетчатке глаза, шванновской оболочке нервных волокон,

Слайд 10Глобоидноклеточная лейкодистрофия (болезнь Краббе)
Редкое нарушение, которое поражает миелиновую оболочку нерва и наследуется
Глобоидноклеточная лейкодистрофия (болезнь Краббе)
Редкое нарушение, которое поражает миелиновую оболочку нерва и наследуется

Слайд 11Болезнь Пелицеуса-Мерцбахера
Характеризуется формированием островков интактного миелина в областях с тяжелой демиелинизацией ("леопардов
Болезнь Пелицеуса-Мерцбахера
Характеризуется формированием островков интактного миелина в областях с тяжелой демиелинизацией ("леопардов

Слайд 12Болезнь Канаван
Наследуется по аутосомно-рецессивному типу. Поврежденный ген находится в 17 хромосоме и
Болезнь Канаван
Наследуется по аутосомно-рецессивному типу. Поврежденный ген находится в 17 хромосоме и

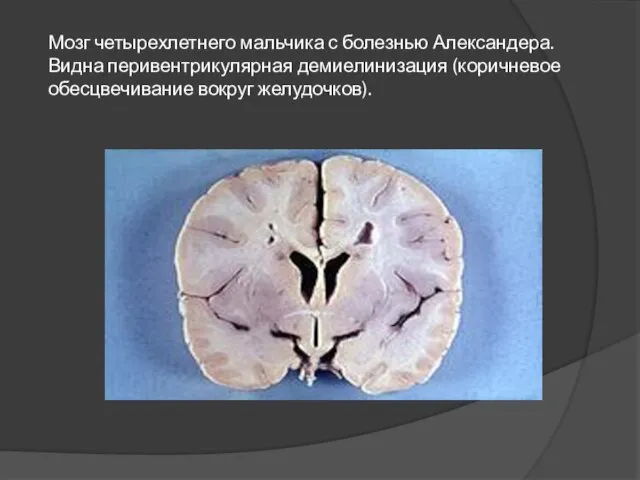
Слайд 13Мозг четырехлетнего мальчика с болезнью Александера. Видна перивентрикулярная демиелинизация (коричневое обесцвечивание вокруг
Мозг четырехлетнего мальчика с болезнью Александера. Видна перивентрикулярная демиелинизация (коричневое обесцвечивание вокруг
Слайд 14Болезнь Александера
Развивается в связи с мутацией в гене, который кодирует глиальный фибриллярный
Болезнь Александера
Развивается в связи с мутацией в гене, который кодирует глиальный фибриллярный

 Магнитогорский государственный технический университет им. Г.И. Носова. Институт строительства архитектуры и искусства. Дизайн
Магнитогорский государственный технический университет им. Г.И. Носова. Институт строительства архитектуры и искусства. Дизайн Царство грибов
Царство грибов О подвигах, о доблестях, о славе
О подвигах, о доблестях, о славе Если не лениться , можно многого добиться
Если не лениться , можно многого добиться Методология Автономного Адаптивного Управлениябионический подход к построению нейроноподобных систем управления
Методология Автономного Адаптивного Управлениябионический подход к построению нейроноподобных систем управления Вставь опущенную букву. Объясни свой выбор.
Вставь опущенную букву. Объясни свой выбор. Тема урока :
Тема урока : Карта развития сотрудника КЦ
Карта развития сотрудника КЦ CASE OF KONSTANTIN MARKIN
CASE OF KONSTANTIN MARKIN Среда МЭО Китая
Среда МЭО Китая Сказка в русской живописи
Сказка в русской живописи Требования в кабинету информатики. Подготовили: студенты группы М-5-В Тарасенко А.А., Тарасенко А.В., Салахова Л.А., Чуйко И.В.
Требования в кабинету информатики. Подготовили: студенты группы М-5-В Тарасенко А.А., Тарасенко А.В., Салахова Л.А., Чуйко И.В. Презентация на тему Химия в быту
Презентация на тему Химия в быту  Тайм-менеджмент: поиск и восстановление личностных ресурсов
Тайм-менеджмент: поиск и восстановление личностных ресурсов Презентация на тему Горы России
Презентация на тему Горы России Испьзование Sanako 1200 в учебной среде школы Наброски слушателя семинара
Испьзование Sanako 1200 в учебной среде школы Наброски слушателя семинара В гостях у сказки (театральная деятельность)
В гостях у сказки (театральная деятельность) Проблеми профілактики девіантної поведінки серед неповнолітніх
Проблеми профілактики девіантної поведінки серед неповнолітніх Презентация на тему Лиственные деревья России
Презентация на тему Лиственные деревья России Интерактивные среды на уроке математики
Интерактивные среды на уроке математики Развитие и затухание морских волн
Развитие и затухание морских волн Электронная школа руководителей
Электронная школа руководителей Фёдор Павлович Решетников (1906 — 1988)
Фёдор Павлович Решетников (1906 — 1988) Образец сайта https://ama-vida.com/
Образец сайта https://ama-vida.com/ Introducere în Testare
Introducere în Testare П. А. Столыпин – палач или великий реформатор ?
П. А. Столыпин – палач или великий реформатор ? Шокирующая реклама
Шокирующая реклама Синтаксис и пунктуация. Пунктуационный разбор
Синтаксис и пунктуация. Пунктуационный разбор