- Мониторинг и аудит клинических исследований

Содержание
- 2. Определение Мониторинг - деятельность, заключающаяся в контроле за ходом клинического исследования, обеспечении его проведения, сбора данных
- 3. Мониторирование может осуществляться сотрудник спонсора контрактно-исследовательская организация (Contract Research Organisation, CRO) независимый монитор
- 4. Цели мониторирования Убедиться что: права и благополучие субъектов защищены представленные данные являются точными, полными и подтверждаются
- 5. Виды визитов визит селекции исследовательского центра визит открытия центра плановый мониторинговый визит внеплановый мониторинговый визит ко-мониторинговый
- 6. Обязанности монитора проверить адекватность квалификации исследователей и достаточность имеющегося персонала для проведения исследований проверка соблюдения исследователем
- 7. Обязанности монитора проверить сроки годности и условия хранения препарата убедиться в том, что назначение препаратов сделано
- 8. Аудит клинических исследований
- 9. Контроль качества главный исследователь, исследовательская команда монитор центра (мониторинговый визит) спонсор / КИО (ко-мониторинговый визит) Data
- 10. Определение Аудит - комплексная и независимая проверка относящихся к исследованию деятельности и документации, проводимая для подтверждения
- 11. Цели и задачи аудита Убедиться что: обеспечивается безопасность и соблюдаются права субъектов исследования исследователь и персонал
- 12. Виды аудитов Внутренний – проводится сотрудниками отдела качества фармацевтической компании или КИО, которые непосредственно участвуют в
- 13. Аудит исследовательского центра Плановый – 10-25% центров Вне плановый (For Cause Audit) – критерии выбора: большое
- 14. Время проведения аудита Перед исследованием (обычно для фазы I) В период включения В период наблюдения за
- 15. Объекты проверки Случайно выбранные ИРК 100% информированные согласия 100% несовершеннолетних пациентов 100% серьезные нежелательные явления Регуляторные
- 16. Классификация находок аудита Значительное несоответствие правилам ICH GCP и существенные ошибки в данных исследования – (major
- 17. Инспекция клинических исследований
- 18. Определение Инспекция - действие уполномоченных органов, заключающееся в официальной проверке документации, оборудования, иных материалов, имеющих, по
- 19. Проводится Министерство здравоохранения и социального развития – для исследований, проводящихся на территории Российской Федерации Food and
- 20. Инспекция Министерства здравоохранения и социального развития Инспектируемый центр предупреждается о проверке в письменной форме Объекты проверки:
- 21. FDA инспекция Плановые - центры для проведения плановых инспекций выбираются наугад, когда нет подозрений в каких-либо
- 22. FDA инспекция Внеплановые – критерии выбора: Исследователь набрал запланированное количество пациентов для проведения клинического исследования «слишком
- 23. Объекты инспекции Роль каждого участника исследования, входящего в группу исследователя Степень делегирования полномочий от исследователя. Где
- 24. Классификация результатов NAI — no action indicated. Требования к проведению исследования соблюдены полностью; VAI — voluntary
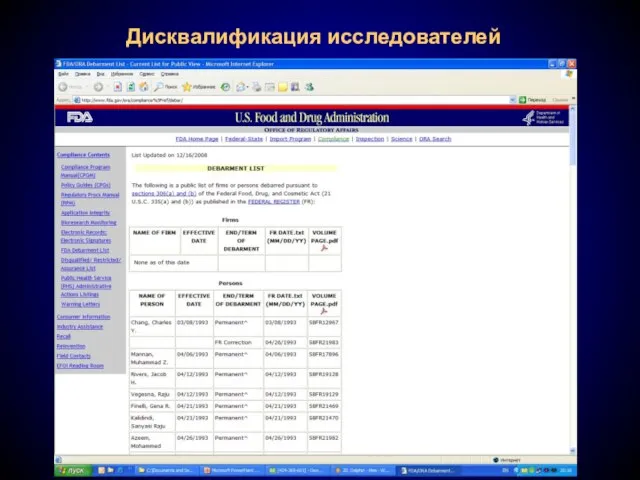
- 25. Дисквалификация исследователей В том случае, если в ходе инспекции будет выявлено, что исследователь неоднократно или намеренно
- 26. Дисквалификация исследователей
- 27. FDA инспекция В течение 15 дней в FDA должен быть послан ответ с планом корректирующих мероприятий
- 29. Скачать презентацию
Слайд 2Определение
Мониторинг - деятельность, заключающаяся в контроле за ходом клинического исследования, обеспечении
Определение
Мониторинг - деятельность, заключающаяся в контроле за ходом клинического исследования, обеспечении

Слайд 3Мониторирование может осуществляться
сотрудник спонсора
контрактно-исследовательская организация (Contract Research Organisation, CRO)
независимый
Мониторирование может осуществляться
сотрудник спонсора
контрактно-исследовательская организация (Contract Research Organisation, CRO)
независимый

Слайд 4Цели мониторирования
Убедиться что:
права и благополучие субъектов защищены
представленные данные являются
Цели мониторирования
Убедиться что:
права и благополучие субъектов защищены
представленные данные являются

Слайд 5Виды визитов
визит селекции исследовательского центра
визит открытия центра
плановый мониторинговый визит
Виды визитов
визит селекции исследовательского центра
визит открытия центра
плановый мониторинговый визит

Слайд 6Обязанности монитора
проверить адекватность квалификации исследователей и достаточность имеющегося персонала для проведения
Обязанности монитора
проверить адекватность квалификации исследователей и достаточность имеющегося персонала для проведения

Слайд 7Обязанности монитора
проверить сроки годности и условия хранения препарата
убедиться в том,
Обязанности монитора
проверить сроки годности и условия хранения препарата
убедиться в том,

Слайд 8Аудит
клинических исследований
Аудит
клинических исследований

Слайд 9Контроль качества
главный исследователь, исследовательская команда
монитор центра (мониторинговый визит)
спонсор /
Контроль качества
главный исследователь, исследовательская команда
монитор центра (мониторинговый визит)
спонсор /

Слайд 10Определение
Аудит - комплексная и независимая проверка относящихся к исследованию деятельности и
Определение
Аудит - комплексная и независимая проверка относящихся к исследованию деятельности и

Слайд 11Цели и задачи аудита
Убедиться что:
обеспечивается безопасность и соблюдаются права субъектов исследования
Цели и задачи аудита
Убедиться что:
обеспечивается безопасность и соблюдаются права субъектов исследования

Слайд 12Виды аудитов
Внутренний – проводится сотрудниками отдела качества фармацевтической компании или КИО, которые
Виды аудитов
Внутренний – проводится сотрудниками отдела качества фармацевтической компании или КИО, которые

Слайд 13Аудит исследовательского центра
Плановый – 10-25% центров
Вне плановый (For Cause Audit) – критерии
Аудит исследовательского центра
Плановый – 10-25% центров
Вне плановый (For Cause Audit) – критерии

Слайд 14Время проведения аудита
Перед исследованием (обычно для фазы I)
В период включения
Время проведения аудита
Перед исследованием (обычно для фазы I)
В период включения

Слайд 15Объекты проверки
Случайно выбранные ИРК
100% информированные согласия
100% несовершеннолетних пациентов
100%
Объекты проверки
Случайно выбранные ИРК
100% информированные согласия
100% несовершеннолетних пациентов
100%

Слайд 16Классификация находок аудита
Значительное несоответствие правилам ICH GCP и существенные ошибки в
Классификация находок аудита
Значительное несоответствие правилам ICH GCP и существенные ошибки в

Слайд 17Инспекция
клинических исследований
Инспекция
клинических исследований

Слайд 18Определение
Инспекция - действие уполномоченных органов, заключающееся в официальной проверке документации, оборудования,
Определение
Инспекция - действие уполномоченных органов, заключающееся в официальной проверке документации, оборудования,

Слайд 19Проводится
Министерство здравоохранения и социального развития – для исследований, проводящихся на территории
Проводится
Министерство здравоохранения и социального развития – для исследований, проводящихся на территории

Слайд 20Инспекция Министерства здравоохранения и
социального развития
Инспектируемый центр предупреждается о проверке в письменной
Инспекция Министерства здравоохранения и
социального развития
Инспектируемый центр предупреждается о проверке в письменной

Слайд 21FDA инспекция
Плановые - центры для проведения плановых инспекций выбираются наугад, когда нет
FDA инспекция
Плановые - центры для проведения плановых инспекций выбираются наугад, когда нет

Слайд 22FDA инспекция
Внеплановые – критерии выбора:
Исследователь набрал запланированное количество пациентов для проведения клинического
FDA инспекция
Внеплановые – критерии выбора:
Исследователь набрал запланированное количество пациентов для проведения клинического

Слайд 23Объекты инспекции
Роль каждого участника исследования, входящего в группу исследователя
Степень делегирования
Объекты инспекции
Роль каждого участника исследования, входящего в группу исследователя
Степень делегирования

Слайд 24Классификация результатов
NAI — no action indicated. Требования к проведению исследования соблюдены полностью;
VAI
Классификация результатов
NAI — no action indicated. Требования к проведению исследования соблюдены полностью;
VAI

Слайд 25Дисквалификация исследователей
В том случае, если в ходе инспекции будет выявлено, что исследователь
Дисквалификация исследователей
В том случае, если в ходе инспекции будет выявлено, что исследователь

Слайд 26Дисквалификация исследователей
Дисквалификация исследователей
Слайд 27FDA инспекция
В течение 15 дней в FDA должен быть послан ответ с
FDA инспекция
В течение 15 дней в FDA должен быть послан ответ с

 Введение в тактику
Введение в тактику  «Готовимся к проверке ФСС в связи с новыми правилами заполнения нового бланка больничного»
«Готовимся к проверке ФСС в связи с новыми правилами заполнения нового бланка больничного» Onega — city by the sea
Onega — city by the sea Самая долгая война в истории
Самая долгая война в истории ИТОГИ КОНТРОЛЬНО-НАДЗОРНОЙ ДЕЯТЕЛЬНОСТИ В СФЕРЕ ВЕТЕРИНАРИИ за 2011 год
ИТОГИ КОНТРОЛЬНО-НАДЗОРНОЙ ДЕЯТЕЛЬНОСТИ В СФЕРЕ ВЕТЕРИНАРИИ за 2011 год Молодежная вечеринка
Молодежная вечеринка Калининградская область
Калининградская область А.С.Пушкин в Лицее
А.С.Пушкин в Лицее Презентация на тему Славные символы России
Презентация на тему Славные символы России Капля йода в бочке меда
Капля йода в бочке меда Венецианское биенале 2016: ахитектура социальной ответственности
Венецианское биенале 2016: ахитектура социальной ответственности Presentation title goes here Subtitle goes here
Presentation title goes here Subtitle goes here  Универсальные учебные действия - умение учится, способность к саморазвитию
Универсальные учебные действия - умение учится, способность к саморазвитию Об опьяняющих свойствах спиртных напитков люди узнали не менее чем за 8000 лет до нашей эры.
Об опьяняющих свойствах спиртных напитков люди узнали не менее чем за 8000 лет до нашей эры. Военная форма и знаки различия в России
Военная форма и знаки различия в России ПЕДАГОГИЧЕСКИЙ СОВЕТ Развитие учительского потенциала как одно из требований модернизации образования 27.03.12.
ПЕДАГОГИЧЕСКИЙ СОВЕТ Развитие учительского потенциала как одно из требований модернизации образования 27.03.12. Что значит таинственное слово "Батик"?
Что значит таинственное слово "Батик"? Бизнес-проект. Финансирование. Уставный капитал. Кредиты. Инвестиции. Распределение прибыли
Бизнес-проект. Финансирование. Уставный капитал. Кредиты. Инвестиции. Распределение прибыли Бавкаева Булгун Доржиевна
Бавкаева Булгун Доржиевна Профессиональная деятельность учителя русского языка и литературы МОУ «Сернурская средняя (полная) общеобразовательная школа №2
Профессиональная деятельность учителя русского языка и литературы МОУ «Сернурская средняя (полная) общеобразовательная школа №2 Школьная летняя площадка. Творческий август
Школьная летняя площадка. Творческий август Организационная схема проведения ЕГЭ в 2012 году
Организационная схема проведения ЕГЭ в 2012 году УНИВЕРСИТЕТСКАЯ БИБЛИОТЕКА В ИНФОРМАЦИОННО-ОБРАЗОВАТЕЛЬНОЙ СРЕДЕ Опарина О. Д., г. Екатеринбург, Уральский государственный униве
УНИВЕРСИТЕТСКАЯ БИБЛИОТЕКА В ИНФОРМАЦИОННО-ОБРАЗОВАТЕЛЬНОЙ СРЕДЕ Опарина О. Д., г. Екатеринбург, Уральский государственный униве Платность расчетных услуг системы БЭСП.- плата за оказание расчетных услуг по переводу платежей - плата за информационные услуг
Платность расчетных услуг системы БЭСП.- плата за оказание расчетных услуг по переводу платежей - плата за информационные услуг Коммерческая деятельность розничной торговой организации по формированию ассортимента товаров и пути ее совершенствования
Коммерческая деятельность розничной торговой организации по формированию ассортимента товаров и пути ее совершенствования Хохломская роспись. Хохлома
Хохломская роспись. Хохлома Робот Экипаж
Робот Экипаж Презентация на тему Свойства логарифмов (10 класс)
Презентация на тему Свойства логарифмов (10 класс)