- Наследственные и врожденные формы патологии

Содержание
- 2. Патогенез наследственных болезней В результате мутаций образуется аномальный ген с измененным кодом. ! ! Реализация действия
- 3. Реализация действия аномального гена Первый путь: аномальный ген, утративший код синтеза структурного или функционально важного белка
- 4. Болезни, возникающие по первому патогенетическому варианту • гипоальбуминемия — ↓ количества альбуминов в крови, что предрасполагает
- 5. Дальтонизм (цветовая слепота) – неспособность различать красный и зелёный цвет, красный и синий или синий и
- 6. Многие люди с нарушением цветовосприятия не увидят на этом изображении число 83 Люди с протанопией не
- 7. Гемофилия появляется из-за изменения одного гена в хромосоме X. Различают три типа гемофилии (A, B, C).
- 8. Гемофилия - связанное с нарушением коагуляции (процессом свёртывания крови). При этом заболевании возникают кровоизлияния в суставы,
- 9. болезнь на сегодняшний день неизлечима Обычно болезнью страдают мужчины (наследование, сцепленное с полом), женщины же выступают
- 10. Реализация действия аномального гена Второй путь: аномальный ген, утративший код нормальной программы синтеза фермента ► прекращение
- 11. Болезни, возникающие по второму патогенетическому пути Фенилкетонурия — наиболее частая форма возникает в результате мутации структурного
- 12. Проявляется в виде нарушения умственного развития, дефектом нервной системы у новорожденных. Это заболевание успешно лечится специально
- 14. Альбинизм. Причина → недостаток фермента тирозиназы в меланоцитах — клетках, синтезирующих пигмент меланин. При отсутствии меланина
- 15. ЛЕЧЕНИЕ БЕЗУСПЕШНО!
- 16. Реализация действия аномального гена Третий путь ⇒ аномальный ген с патологическим кодом ► синтез патологической информационной
- 17. Серповидно-клеточная анемия ☞ синтез патологического S-гемоглобина, отличающегося от нормального Hb тем, что в 6-м положении β-полипептидной
- 18. Нормальные и деформированные эритроциты в крови человека
- 19. Муковисцидоз Кистозный фиброз является одним из самых распространенных моногенных заболеваний в Европе (1 на 2500 новорожденных).
- 20. Патологический ген локализуется в середине длинного плеча 7-й хромосомы. Если оба родителя гетерозиготны, то риск рождения
- 21. Муковисцидоз имеет аутосомно-рецессивный тип наследования
- 22. Изменения в поджелудочной железе, органах дыхания, желудочно-кишечном тракте регистрируются уже во внутриутробном периоде и с возрастом
- 23. Прогноз при муковисцидозе до настоящего времени остается серьёзным. Летальность составляет 50—60 %, среди детей раннего возраста
- 26. Симптом барабанных палочек и часовых стекол при муковисцидозе.
- 27. Хромосомные болезни Являются особым видом наследственной патологии, связанной с повреждением их структуры (хромосомными мутациями) или нарушением
- 28. Особенности течения и проявления хромосомных болезней Общим для всех форм хромосомных болезней является множественность поражения: черепно-лицевые
- 29. Синдром Да́уна (трисомия по 21 хромосоме ) — одна из форм геномной патологии, при которой чаще
- 32. На ладони часто обнаруживают поперечную складку Болезнь Дауна Кариотип больного
- 34. Кариотип при транслокационном синдроме Дауна (одна 21-я хромосома присоединена к 15-й хромосоме — указано стрелкой) Женщина
- 35. Наиболее частые внешние признаки синдрома Дауна
- 36. Врождённые пороки внутренних органов, сниженная приспособленность детей с синдромом Дауна часто приводят к летальному исходу в
- 37. Диагностика: В периоде внутриутробного развития синдрома Дауна может быть выявлен при проведении биохимического скрининга беременных во
- 38. В последнее время внимание генетиков привлечено к изучению Х-сцепленной умственной отсталости (синдром ломкой, или фрагильной Х-хромосомы,
- 39. После выявления этой наследственной формы умственной отсталости стала понятной большая частота встречаемости интеллектуального недоразвития у мальчиков.
- 40. Речь изобилует повторами, часто встречается своеобразное заикание (логоневроз). Для детей характерна двигательная расторможенность и некоторые симптомы
- 41. Аномалии соматических хромосом Трисомия по 18-й паре — синдром Эдвардса (частота 1:7000). Дети рождаются слабыми, имеют
- 42. Синдром Э́двардса (синдром трисомии 18) характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. (трисомия 18;
- 43. Синдром трисомии 18 (синдром Эдвардса). А — внешний вид больного; Б — кариотип больного при трисомии
- 44. Прогноз: Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3
- 45. Для подтверждения диагноза синдрома Эдвардса и дальнейшего медико-генетического прогнозирования состояния здоровья будущего потомства родителей показано пренатальное
- 46. Аномалии соматических хромосом Трисомии по 8,9,13-й паре ☞синдром Патау (1:6000). Дети очень рано гибнут (96 %
- 47. Синдром Патау Частота синдрома Патау среди новорождённых равна 1:5000-1:7000. Цитогенетические варианты этого синдрома следующие. Простая полная
- 48. Виды внутрихромосомиых перестроек: 1 — исходная пара гомологичных хромосом; 2 — потеря участка DEFH хромосомы (делеция);
- 49. Синдром трисомии 13 (синдром Патау). А — внешний вид больного; Б — кариотип больного с трисомией
- 50. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25-30% ниже средних величин), которую нельзя
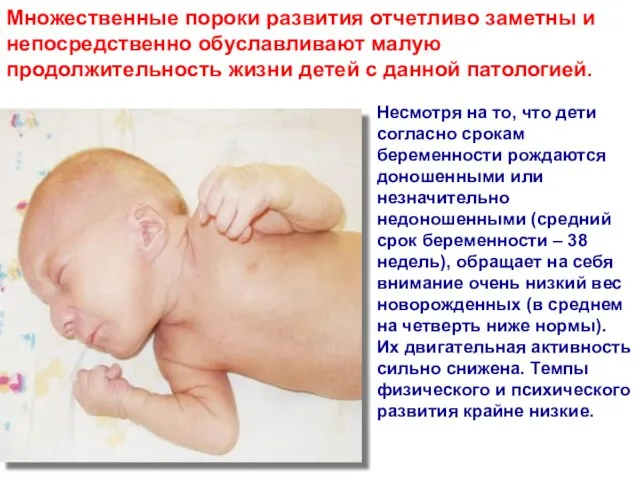
- 51. Множественные пороки развития отчетливо заметны и непосредственно обуславливают малую продолжительность жизни детей с данной патологией. Несмотря
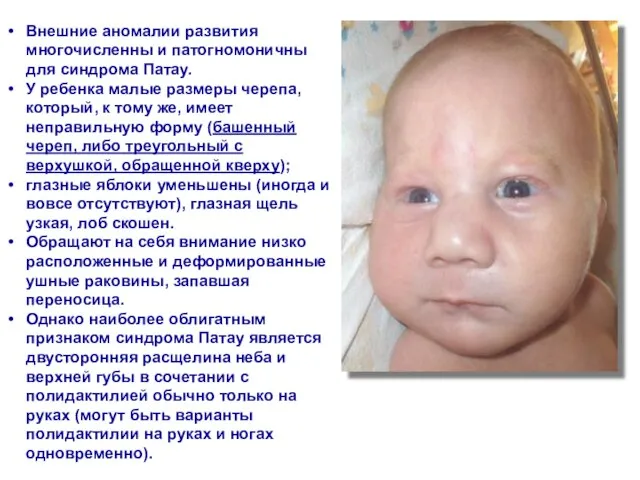
- 52. Внешние аномалии развития многочисленны и патогномоничны для синдрома Патау. У ребенка малые размеры черепа, который, к
- 53. В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели
- 54. Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с
- 55. Аномалии, связанные с половыми хромосомами
- 56. Синдром дубль-Y Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г. Частота синдрома
- 57. 47, XYY
- 58. 47, XYY
- 59. Синдром Кляйнфельтера Встречается у мужчин. Частота 2 на 1000 новорожденных мальчиков. Кариотип чаще: XXY, общее количество
- 60. Для мужчин с синдромом Клайнфельтера характерны высокий рост, длинные конечности и относительно короткое туловище, евнухоидизм, бесплодие,
- 65. Синдром Шерешевского Тернера - возникает при слиянии патологической женской гаметы, лишенной Х-хромосомы, с нормальной мужской гаметой,
- 66. Впервые эта болезнь как наследственная была описана в 1925 г. Н.А. Шерешевским, который считал, что она
- 67. При синдроме Тернера половые железы обычно представляют собой недифференцированные соединительнотканные тяжи, не содержащие элементов гонад. Реже
- 68. Для течения постнатального периода характерно общее беспокойство новорожденных, нарушение сосательного рефлекса, срыгивания фонтаном, рвота. В раннем
- 73. Прогноз для жизни при с. Ш.-Т. благоприятный, исключение составляют больные с тяжелыми врожденными пороками сердца и
- 74. Синдром трисомии X. Встречается у женщин (1:1000), кариотип — 47 (XXX). В неделящихся клетках видны два
- 75. 47 (XXX)
- 76. 47 (XXX)
- 78. Ядро клетки самки. Наверху: при помощи FISH определяются обе Х-хромосомы. Внизу: окрашивание ДНК. Тельце Барра показано
- 79. Аномалии хромосом
- 80. Синдром Кошачьего крика - объясняется частичной моносомией. Развивается при делеции (с утратой от трети до половины,
- 83. Подходы в борьбе с наследственными болезнями 1. Массовое «просеивание» новорожденных на наследственные дефекты обмена веществ (выявление
- 85. Скачать презентацию
Слайд 2Патогенез наследственных болезней
В результате мутаций образуется аномальный ген с измененным кодом.
Патогенез наследственных болезней
В результате мутаций образуется аномальный ген с измененным кодом.

Слайд 3Реализация действия аномального гена
Первый путь: аномальный ген, утративший код синтеза структурного
Реализация действия аномального гена
Первый путь: аномальный ген, утративший код синтеза структурного

Слайд 4Болезни, возникающие по первому патогенетическому варианту
• гипоальбуминемия — ↓ количества альбуминов в
Болезни, возникающие по первому патогенетическому варианту
• гипоальбуминемия — ↓ количества альбуминов в

Слайд 5Дальтонизм (цветовая слепота) – неспособность различать красный и зелёный цвет, красный и
Дальтонизм (цветовая слепота) – неспособность различать красный и зелёный цвет, красный и

Слайд 6Многие люди с нарушением цветовосприятия не увидят на этом изображении число 83
Люди
Многие люди с нарушением цветовосприятия не увидят на этом изображении число 83
Люди
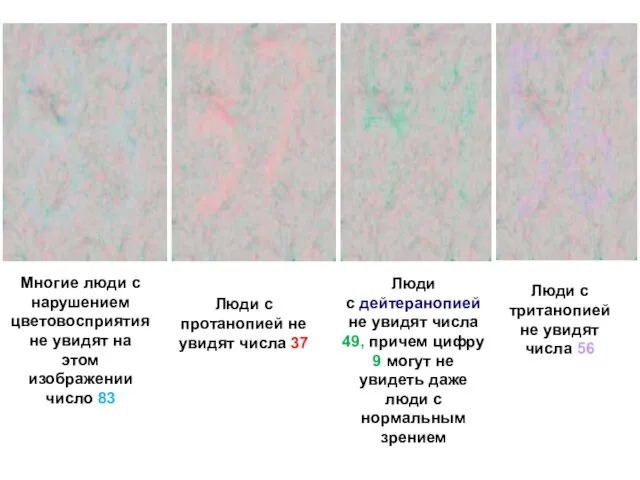
Слайд 7Гемофилия появляется из-за изменения одного гена в хромосоме X. Различают три типа
Гемофилия появляется из-за изменения одного гена в хромосоме X. Различают три типа

Слайд 8Гемофилия - связанное с нарушением коагуляции (процессом свёртывания крови).
При этом заболевании возникают
Гемофилия - связанное с нарушением коагуляции (процессом свёртывания крови).
При этом заболевании возникают

Слайд 9болезнь на сегодняшний день неизлечима
Обычно болезнью страдают мужчины (наследование, сцепленное с полом),
болезнь на сегодняшний день неизлечима
Обычно болезнью страдают мужчины (наследование, сцепленное с полом),

Слайд 10Реализация действия аномального гена
Второй путь: аномальный ген, утративший код нормальной программы
Реализация действия аномального гена
Второй путь: аномальный ген, утративший код нормальной программы

Слайд 11Болезни, возникающие по второму патогенетическому пути
Фенилкетонурия — наиболее частая форма возникает в
Болезни, возникающие по второму патогенетическому пути
Фенилкетонурия — наиболее частая форма возникает в

Слайд 12Проявляется в виде нарушения умственного развития, дефектом нервной системы у новорожденных.
Это заболевание
Проявляется в виде нарушения умственного развития, дефектом нервной системы у новорожденных.
Это заболевание


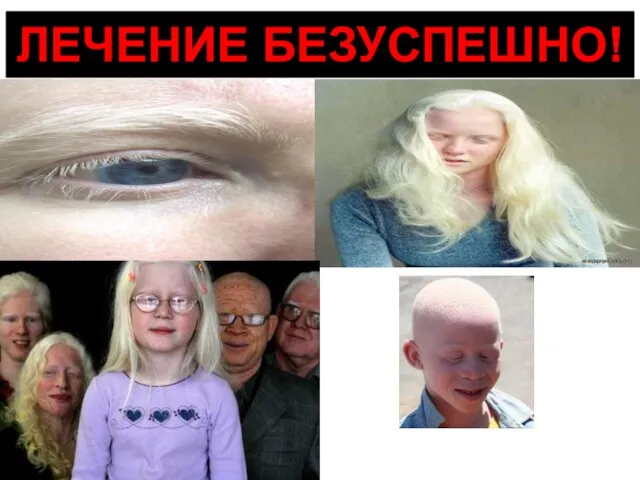
Слайд 14Альбинизм. Причина → недостаток фермента тирозиназы в меланоцитах — клетках, синтезирующих пигмент
Альбинизм. Причина → недостаток фермента тирозиназы в меланоцитах — клетках, синтезирующих пигмент

Слайд 15ЛЕЧЕНИЕ БЕЗУСПЕШНО!
ЛЕЧЕНИЕ БЕЗУСПЕШНО!
Слайд 16 Реализация действия аномального гена
Третий путь ⇒ аномальный ген с патологическим
Реализация действия аномального гена
Третий путь ⇒ аномальный ген с патологическим

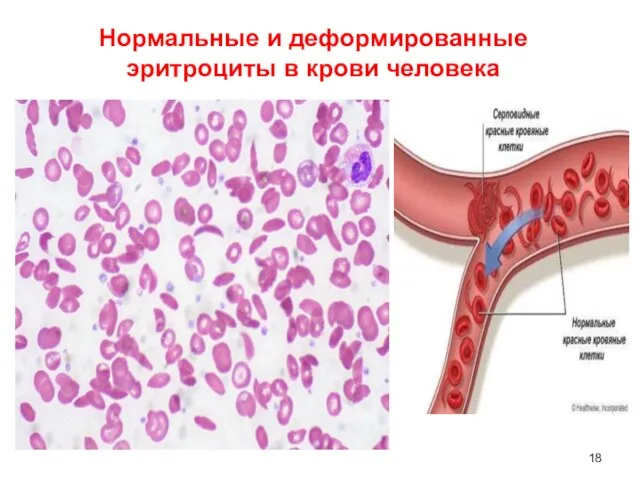
Слайд 17Серповидно-клеточная анемия ☞ синтез патологического S-гемоглобина, отличающегося от нормального Hb тем, что
Серповидно-клеточная анемия ☞ синтез патологического S-гемоглобина, отличающегося от нормального Hb тем, что

Слайд 18Нормальные и деформированные эритроциты в крови человека
Нормальные и деформированные эритроциты в крови человека
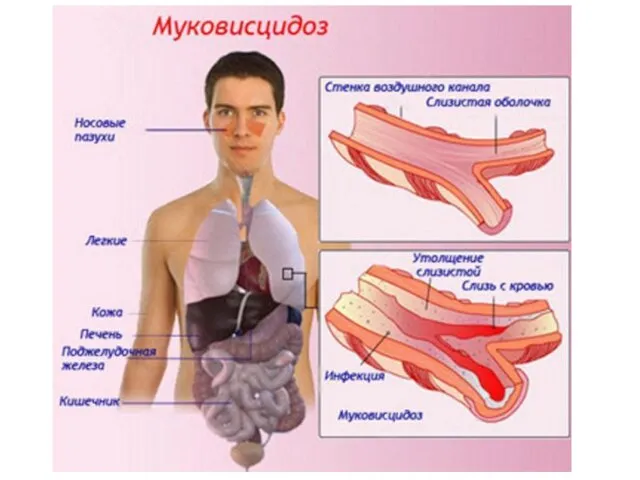
Слайд 19Муковисцидоз
Кистозный фиброз является одним из самых распространенных моногенных заболеваний в Европе (1
Муковисцидоз
Кистозный фиброз является одним из самых распространенных моногенных заболеваний в Европе (1

Слайд 20Патологический ген локализуется в середине длинного плеча 7-й хромосомы.
Если оба родителя
Патологический ген локализуется в середине длинного плеча 7-й хромосомы.
Если оба родителя

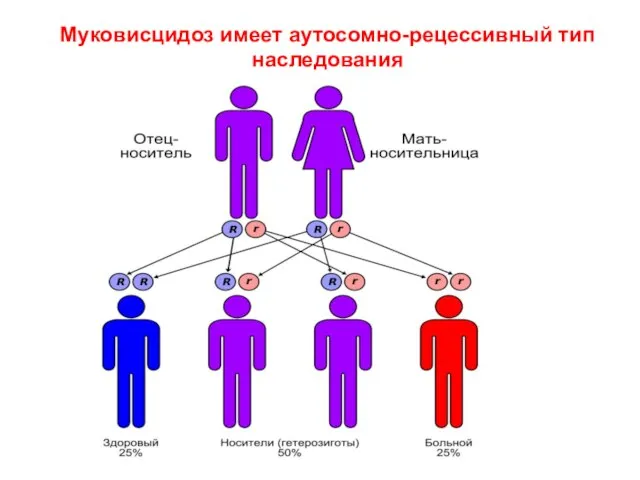
Слайд 21Муковисцидоз имеет аутосомно-рецессивный тип наследования
Муковисцидоз имеет аутосомно-рецессивный тип наследования
Слайд 22Изменения в поджелудочной железе, органах дыхания, желудочно-кишечном тракте регистрируются уже во внутриутробном
Изменения в поджелудочной железе, органах дыхания, желудочно-кишечном тракте регистрируются уже во внутриутробном

Слайд 23Прогноз при муковисцидозе до настоящего времени остается серьёзным.
Летальность составляет 50—60 %, среди
Прогноз при муковисцидозе до настоящего времени остается серьёзным.
Летальность составляет 50—60 %, среди


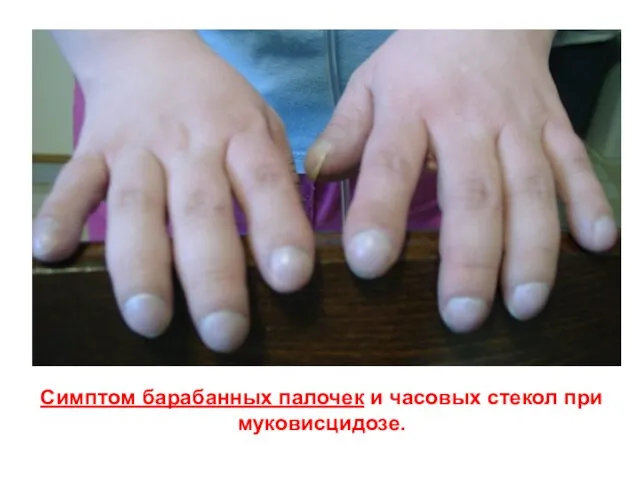
Слайд 26Симптом барабанных палочек и часовых стекол при муковисцидозе.
Симптом барабанных палочек и часовых стекол при муковисцидозе.
Слайд 27
Хромосомные болезни
Являются особым видом наследственной патологии, связанной с повреждением их структуры
Хромосомные болезни
Являются особым видом наследственной патологии, связанной с повреждением их структуры

Слайд 28Особенности течения и проявления хромосомных болезней
Общим для всех форм хромосомных болезней является
Особенности течения и проявления хромосомных болезней
Общим для всех форм хромосомных болезней является

Слайд 29Синдром Да́уна (трисомия по 21 хромосоме ) — одна из форм геномной
Синдром Да́уна (трисомия по 21 хромосоме ) — одна из форм геномной



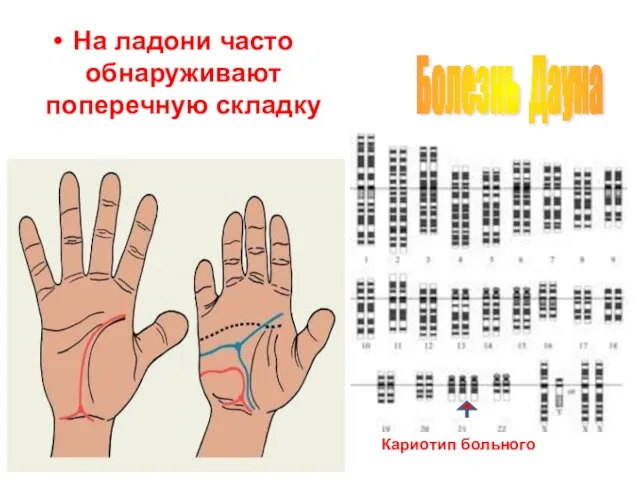
Слайд 32На ладони часто обнаруживают поперечную складку
Болезнь Дауна
Кариотип больного
На ладони часто обнаруживают поперечную складку
Болезнь Дауна
Кариотип больного

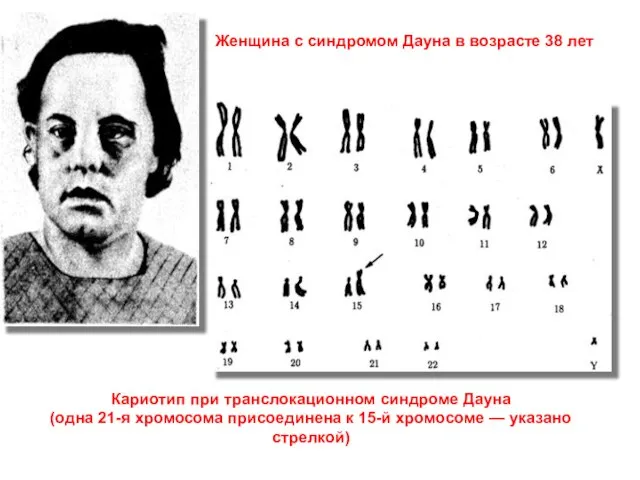
Слайд 34Кариотип при транслокационном синдроме Дауна
(одна 21-я хромосома присоединена к 15-й хромосоме —
Кариотип при транслокационном синдроме Дауна
(одна 21-я хромосома присоединена к 15-й хромосоме —
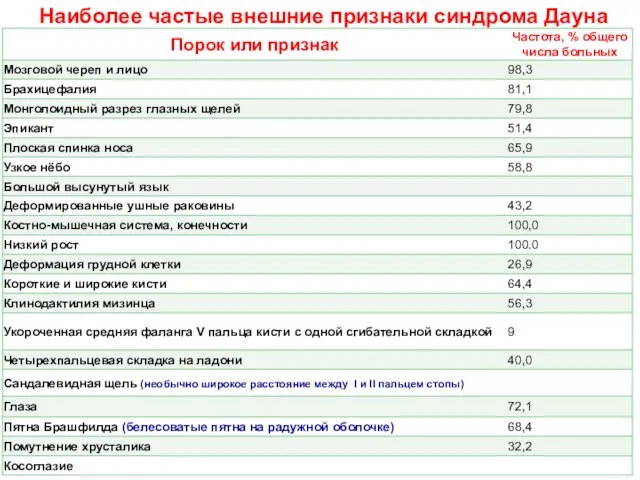
Слайд 35Наиболее частые внешние признаки синдрома Дауна
Наиболее частые внешние признаки синдрома Дауна
Слайд 36Врождённые пороки внутренних органов, сниженная приспособленность детей с синдромом Дауна часто приводят
Врождённые пороки внутренних органов, сниженная приспособленность детей с синдромом Дауна часто приводят

Слайд 37Диагностика:
В периоде внутриутробного развития синдрома Дауна может быть выявлен при проведении биохимического
Диагностика:
В периоде внутриутробного развития синдрома Дауна может быть выявлен при проведении биохимического

Слайд 38В последнее время внимание генетиков привлечено к изучению Х-сцепленной умственной отсталости (синдром
В последнее время внимание генетиков привлечено к изучению Х-сцепленной умственной отсталости (синдром

Слайд 39После выявления этой наследственной формы умственной отсталости стала понятной большая частота встречаемости
После выявления этой наследственной формы умственной отсталости стала понятной большая частота встречаемости

Слайд 40Речь изобилует повторами, часто встречается своеобразное заикание (логоневроз).
Для детей характерна двигательная
Речь изобилует повторами, часто встречается своеобразное заикание (логоневроз).
Для детей характерна двигательная

Слайд 41Аномалии соматических хромосом
Трисомия по 18-й паре — синдром Эдвардса (частота 1:7000).
Аномалии соматических хромосом
Трисомия по 18-й паре — синдром Эдвардса (частота 1:7000).

Слайд 42Синдром Э́двардса (синдром трисомии 18)
характеризуется комплексом множественных пороков развития и трисомией 18
Синдром Э́двардса (синдром трисомии 18)
характеризуется комплексом множественных пороков развития и трисомией 18

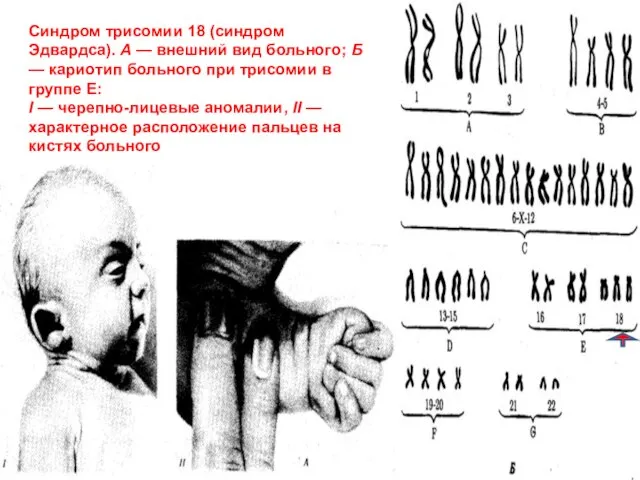
Слайд 43Синдром трисомии 18 (синдром Эдвардса). А — внешний вид больного; Б —
Синдром трисомии 18 (синдром Эдвардса). А — внешний вид больного; Б —
Слайд 44Прогноз: Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в
Прогноз: Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в

Слайд 45Для подтверждения диагноза синдрома Эдвардса и дальнейшего медико-генетического прогнозирования состояния здоровья будущего
Для подтверждения диагноза синдрома Эдвардса и дальнейшего медико-генетического прогнозирования состояния здоровья будущего

Слайд 46Аномалии соматических хромосом
Трисомии по 8,9,13-й паре ☞синдром Патау (1:6000). Дети очень
Аномалии соматических хромосом
Трисомии по 8,9,13-й паре ☞синдром Патау (1:6000). Дети очень

Слайд 47Синдром Патау
Частота синдрома Патау среди новорождённых равна 1:5000-1:7000.
Цитогенетические варианты этого синдрома
Синдром Патау
Частота синдрома Патау среди новорождённых равна 1:5000-1:7000.
Цитогенетические варианты этого синдрома

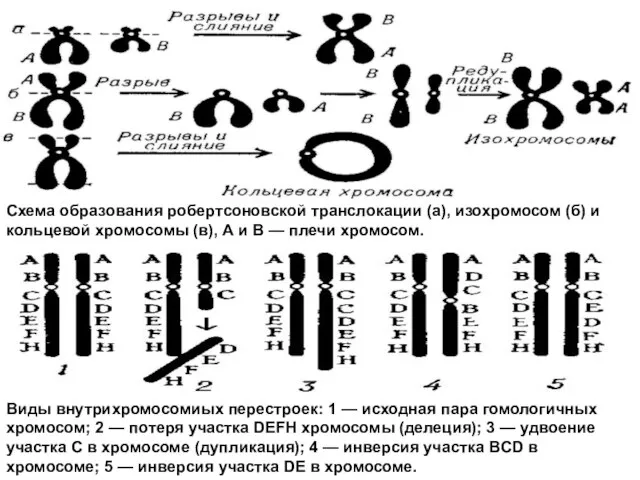
Слайд 48Виды внутрихромосомиых перестроек: 1 — исходная пара гомологичных хромосом; 2 — потеря
Виды внутрихромосомиых перестроек: 1 — исходная пара гомологичных хромосом; 2 — потеря
Слайд 49Синдром трисомии 13 (синдром Патау).
А — внешний вид больного; Б — кариотип
Синдром трисомии 13 (синдром Патау).
А — внешний вид больного; Б — кариотип

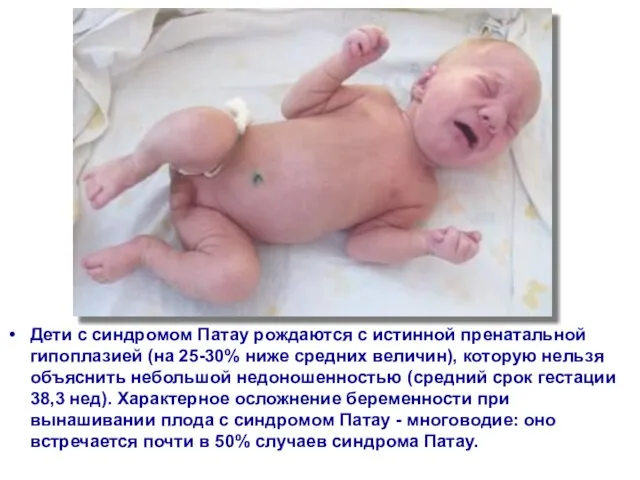
Слайд 50Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25-30% ниже
Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25-30% ниже
Слайд 51Множественные пороки развития отчетливо заметны и непосредственно обуславливают малую продолжительность жизни детей
Множественные пороки развития отчетливо заметны и непосредственно обуславливают малую продолжительность жизни детей
Слайд 52Внешние аномалии развития многочисленны и патогномоничны для синдрома Патау.
У ребенка малые
Внешние аномалии развития многочисленны и патогномоничны для синдрома Патау.
У ребенка малые
Слайд 53В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау

Слайд 54Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по
Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по

Слайд 55Аномалии, связанные с половыми хромосомами
Аномалии, связанные с половыми хромосомами

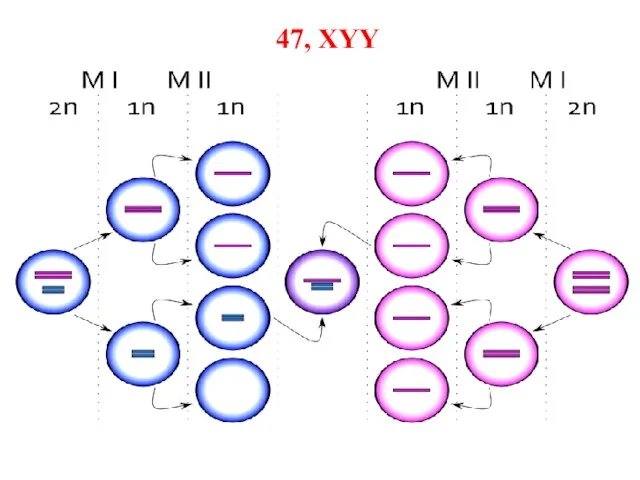
Слайд 56Синдром дубль-Y
Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г.
Синдром дубль-Y
Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г.

Слайд 5747, XYY
47, XYY
Слайд 5847, XYY
47, XYY

Слайд 59Синдром Кляйнфельтера
Встречается у мужчин. Частота 2 на 1000 новорожденных мальчиков. Кариотип чаще:
Синдром Кляйнфельтера
Встречается у мужчин. Частота 2 на 1000 новорожденных мальчиков. Кариотип чаще:

Слайд 60Для мужчин с синдромом Клайнфельтера характерны высокий рост, длинные конечности и относительно
Для мужчин с синдромом Клайнфельтера характерны высокий рост, длинные конечности и относительно





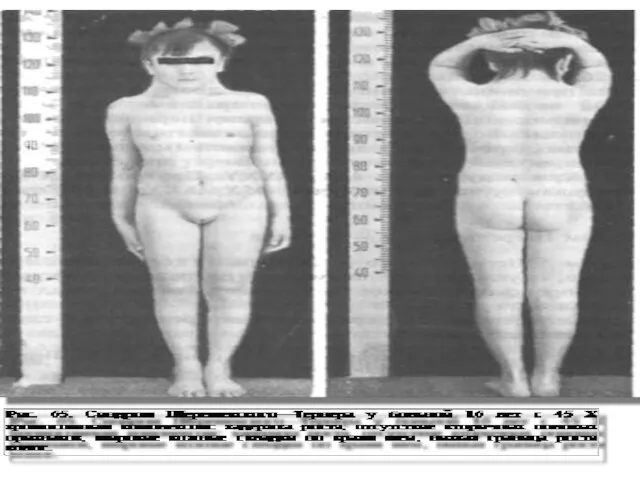
Слайд 65Синдром Шерешевского Тернера - возникает при слиянии патологической женской гаметы, лишенной Х-хромосомы,
Синдром Шерешевского Тернера - возникает при слиянии патологической женской гаметы, лишенной Х-хромосомы,

Слайд 66Впервые эта болезнь как наследственная была описана в 1925 г. Н.А. Шерешевским,
Впервые эта болезнь как наследственная была описана в 1925 г. Н.А. Шерешевским,

Слайд 67При синдроме Тернера половые железы обычно представляют собой недифференцированные соединительнотканные тяжи, не
При синдроме Тернера половые железы обычно представляют собой недифференцированные соединительнотканные тяжи, не

Слайд 68Для течения постнатального периода характерно общее беспокойство новорожденных, нарушение сосательного рефлекса, срыгивания
Для течения постнатального периода характерно общее беспокойство новорожденных, нарушение сосательного рефлекса, срыгивания

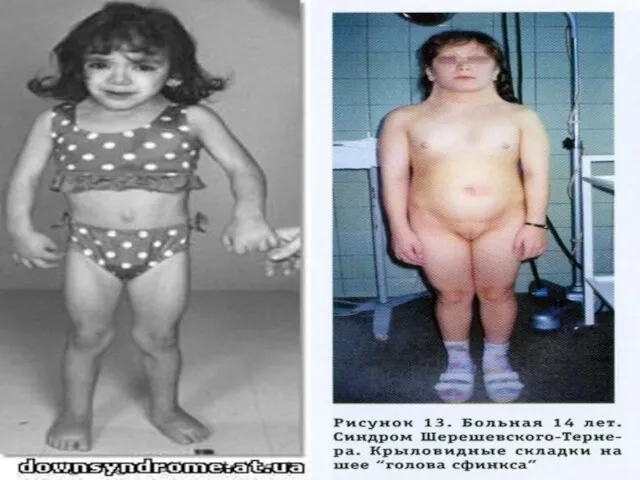


Слайд 73Прогноз для жизни при с. Ш.-Т. благоприятный, исключение составляют больные с тяжелыми
Прогноз для жизни при с. Ш.-Т. благоприятный, исключение составляют больные с тяжелыми

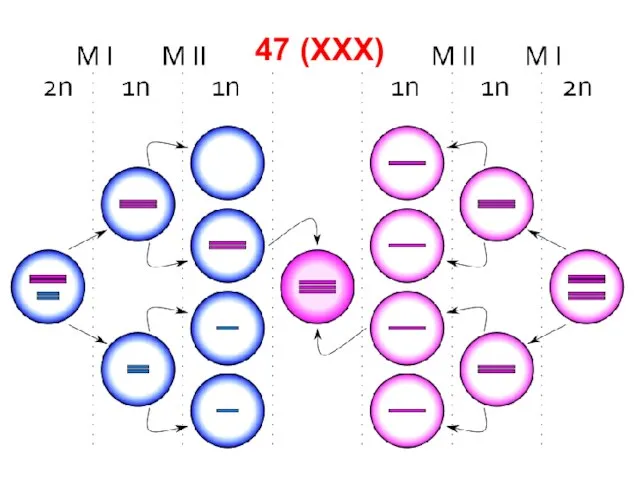
Слайд 74Синдром трисомии X.
Встречается у женщин (1:1000), кариотип — 47 (XXX).
В
Синдром трисомии X.
Встречается у женщин (1:1000), кариотип — 47 (XXX).
В

Слайд 7547 (XXX)
47 (XXX)
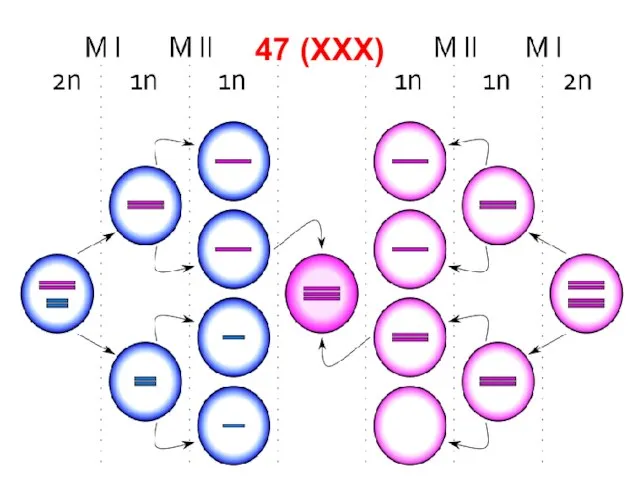
Слайд 7647 (XXX)
47 (XXX)

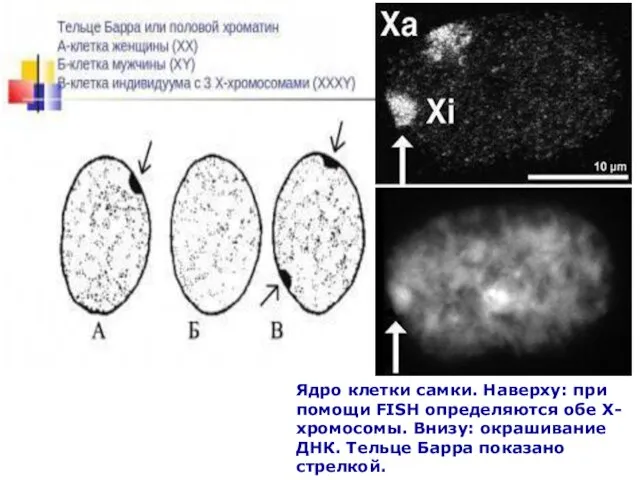
Слайд 78Ядро клетки самки. Наверху: при помощи FISH определяются обе Х-хромосомы. Внизу: окрашивание
Ядро клетки самки. Наверху: при помощи FISH определяются обе Х-хромосомы. Внизу: окрашивание
Слайд 79Аномалии хромосом
Аномалии хромосом

Слайд 80Синдром Кошачьего крика - объясняется частичной моносомией.
Развивается при делеции (с утратой от
Синдром Кошачьего крика - объясняется частичной моносомией.
Развивается при делеции (с утратой от



Слайд 83Подходы в борьбе с наследственными болезнями
1. Массовое «просеивание» новорожденных на наследственные
Подходы в борьбе с наследственными болезнями
1. Массовое «просеивание» новорожденных на наследственные

 Героям Сталинградской битвы
Героям Сталинградской битвы Нефтяная промышленность Тюменской области
Нефтяная промышленность Тюменской области  Диагностические задания для детей
Диагностические задания для детей Общее отчетно-выборное собрание СНТ Рябинушка
Общее отчетно-выборное собрание СНТ Рябинушка Народы России
Народы России Гэрэй амитад
Гэрэй амитад Педагогические условия совершенствования тактики игры в нападении в футболе
Педагогические условия совершенствования тактики игры в нападении в футболе Darkfield Microscope
Darkfield Microscope Физические основы вычислительной техники. Цифровая 3D-медицина
Физические основы вычислительной техники. Цифровая 3D-медицина Презентация на тему Бабушка - Загадушка
Презентация на тему Бабушка - Загадушка Время остановить нельзя, а измерить?
Время остановить нельзя, а измерить? Производство корундовой броне-керамики в АО УАПО
Производство корундовой броне-керамики в АО УАПО Christmas
Christmas Setting Goals and Measuring change
Setting Goals and Measuring change Основы сертификации. Термины и определения. Государственная система сертификации. Тема №6. Занятие №1-5
Основы сертификации. Термины и определения. Государственная система сертификации. Тема №6. Занятие №1-5 Жизнь и судьба наших четвероногих друзей
Жизнь и судьба наших четвероногих друзей Путеводитель Звёздного курса
Путеводитель Звёздного курса ПУБЛИЧНЫЙ ДОКЛАДМОУ Школы №1 г.о. Самара2011г
ПУБЛИЧНЫЙ ДОКЛАДМОУ Школы №1 г.о. Самара2011г Что такое «семья»?
Что такое «семья»? январь 2011 г.
январь 2011 г. Презентация на тему Искусство Итальянского Возрождения
Презентация на тему Искусство Итальянского Возрождения  Средства массовой информации и их виды
Средства массовой информации и их виды Антонио Лучо Вивальди
Антонио Лучо Вивальди  Назначение и состав бурильной колонны
Назначение и состав бурильной колонны Положительные и отрицательные стороны трения
Положительные и отрицательные стороны трения Unit 4 translation
Unit 4 translation Фастфуд Dodo-pizza. Объем выручки с продаж
Фастфуд Dodo-pizza. Объем выручки с продаж Презентация на тему Внешнеэкономические связи ведущих развитых стран
Презентация на тему Внешнеэкономические связи ведущих развитых стран