- Наследственные заболевания

Содержание
- 2. Словарик Хромосомные болезни – наследственные заболевания, обусловленные изменением числа или структуры хромосом. Частота Хромосомных болезней среди
- 3. Моносомия - отсутствие в хромосомном наборе диплоидного организма одной хромосомы. Клетку или организм, у которых та
- 4. Типы наследственности 1. Аутосомно-доминантный тип наследования: а. При достаточном числе потомков признак обнаруживается в каждом поколении
- 5. Наследственные заболевания: Хромосомные болезни Болезни обмена веществ Нарушения иммунитета Болезни с преимущественным поражением эндокринной системы Болезни
- 6. Хромосомные болезни: Синдром Патау Синдром Дауна Синдром Эдвардса Синдром Шершевского-Тернера Синдром «кошачьего крика» Синдром Клайнфельтера Синдром
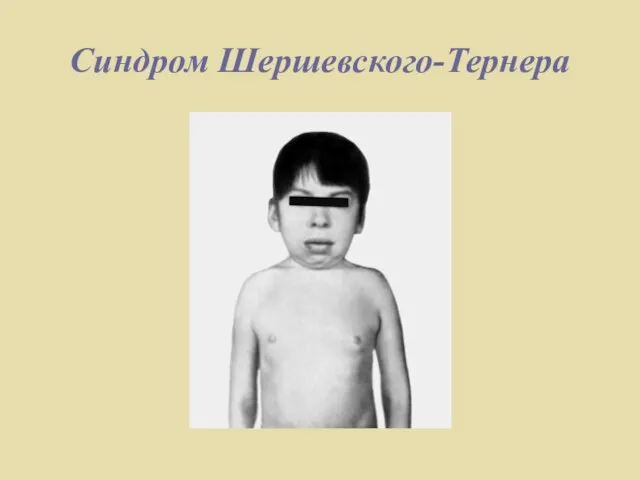
- 7. Синдром Шершевского-Тернера Известно, что пол женщины и мужчины определяется наличием двух половых хромосом: у женщины -
- 8. Синдром Шершевского-Тернера
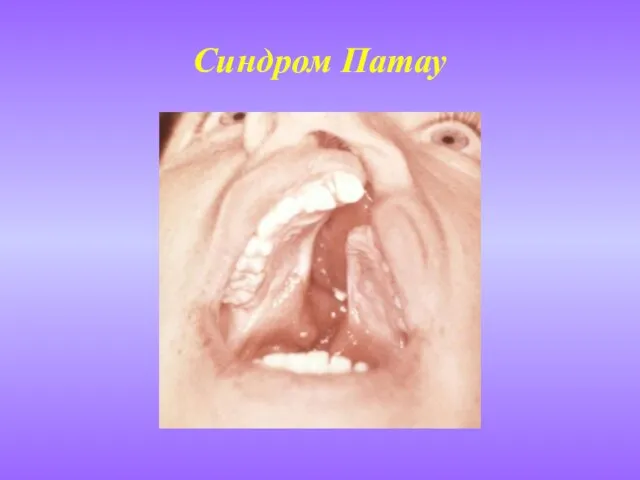
- 9. Синдром Патау (синдром 13-трисомии) Встречается примерно в одном случае на 25 тыс. живорожденных детей. Риск увеличивается
- 10. Синдром Патау
- 11. Синдром Эдвардса (синдром 18-трисомии) Встречается в одном случае на 6600 живорожденных, почти 80% пораженных — девочки.
- 12. Синдром Эдвардса
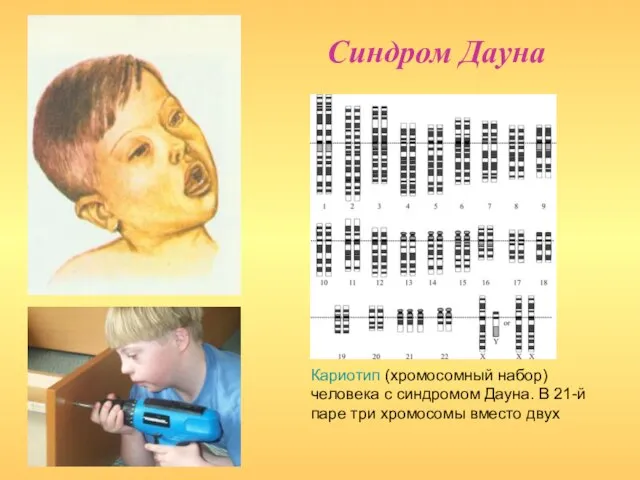
- 13. Синдром Дауна (синдром 21-трисомии) Встречается в одном случае на 7—10 тыс. живорожденных детей обоих полов во
- 14. Синдром Дауна Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три хромосомы вместо двух
- 15. А теперь немного о мало известных хромосомных заболеваниях человека...
- 16. Синдром «кошачьего крика» Синдром кошачьего крика (5р-) обусловлен делецией короткого плеча 5-й хромосомы. Популяционная частота синдрома
- 17. Синдром «кошачьего крика»
- 18. Синдром Клайнфельтера Синдром Клайнфельтера включает случаи полисомии по половым хромосомам, при которых имеется не менее двух
- 19. Синдром Клайнфельтера
- 20. Синдром дубль-Y Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г. Частота синдрома
- 21. Трисомия Х Частота трисомии-Х составляет среди новорожденных девочек и женщин 1:1000, среди умственно отсталых — 0,59
- 22. Болезни обмена веществ: Гомоцистинурия Гистидинемия Нарушения обмена триптофана Глюкоцереброзидозы Финелкетонурия
- 23. Глюкоцереброзидозы Болезнь Гоше – это наиболее часто встречающаяся наследственная болезнь нарушения накопления, при которой имеется дефицит
- 24. Фенилкетонурия Фенилкетонурия (ФКУ) - тяжелое наследственное заболевание, наступающее вследствие врожденного дефекта фермента, отвечающего в организме человека
- 25. Гомоцистинурия Заболевание наследуется по аутосомно-рецессивному типу. Частота гомоцистинурии составляет 1:200 000 новорожденных. В основе заболевания лежит
- 26. Гистидинемия Заболевание возникает в результате отсутствия или недостаточности активности фермента гистидазы. Наследуется аутосомно-рецессивно. Для детей первого
- 27. Нарушения обмена триптофана Болезнь Гартнепа. Аутосомно-рецессивный тип наследования. При данном заболевании наблюдается генетическое изменение транспортной функции
- 28. Нарушения иммунитета Агаммаглобулинемия
- 29. Агаммаглобулинемия АГАММАГЛОБУЛИНЕМИЯ (греч.отриц. приставка а- + гамма-глобулин + греч. haima кровь; син.: пангипогамма-глобулинемия, болезнь Брутона) -
- 30. Болезни с преимущественным поражением эндокринной системы Синдром Бёрьесона Муковисцидоз Кретинизм Лепречаунизм
- 31. Синдром Бёрьесона Бёрьесона-Форсмана-Лемана синдром – наследственная форма слабоумия с ожирением. Психика характеризуется умственным недоразвитием степени идиотии
- 32. Муковисцидоз Муковисцидоз - (от латинского mucus - слизь, viscidus - вязкий) самое распространенное наследственное заболевание, при
- 33. Кретинизм КРЕТИНИЗМ - (от франц. cretin - слабоумный, кретин), эндокринное заболевание - недостаточная функция щитовидной железы,
- 34. Кретинизм На одном из портретов великого Веласкеса изображен Франциско Лескано, неряшливо одетый подросток с крупной головой,
- 35. Лепречаунизм Лепречаунизм (leprechaunismus; ирланд. leprechaun гном; син. Донохью синдром) - наследственная болезнь женщин, обусловленная нарушениями развития
- 36. Болезни крови Гемоглобинопатии Серповидноклеточная анемия Талассемия Гемофилия Тромбофилии
- 37. Гемоглобинопатии Гемоглобинопатии (от гемоглобин и греч. páthos - страдание, болезнь), гемоглобинозы – состояния, обусловленные присутствием в
- 38. Серповидноклеточная анемия Серповидноклеточная анемия (HbS) связана с наличием в эритроцитах патологического гемоглобина S. При этой форме
- 39. Талассемия Талассемия - заболевание, распространённое в средиземноморских странах. Характеризуется значительным повышением содержания HbF в крови. Полагают,
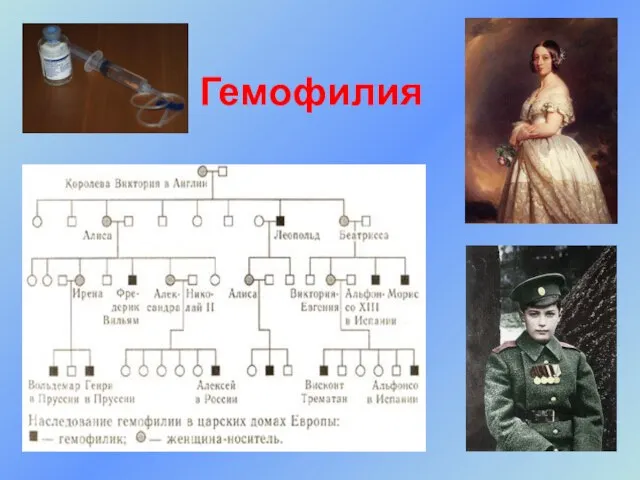
- 40. Гемофилия Гемофилия – Рецессивно наследуемое заболевание, сцепленное с Х- хромосомой. Гемофилия А - нарушение синтеза фактора
- 41. Гемофилия
- 42. Тромбофилии ТРОМБОФИЛИИ ГЕМАТОГЕННЫЕ – состояния, характеризующиеся наклонностью к развитию рецидивирующих тромбозов кровеносных сосудов (преимущественно вен) разной
- 43. Нарушение функций почек Фосфат-диабет Наследственный нефрит
- 44. Фосфат-диабет ФОСФАТ-ДИАБЕТ – доминантно сцепленное с Х-хромосомой заболевание с глубокими нарушениями фосфорно-кальциевого обмена, которые не удается
- 45. Наследственный нефрит НАСЛЕДСТВЕННЫЙ НЕФРИТ. Этиология, патогенез не изучены. Предполагается, что заболевание связано с мутацией гена, контролирующего
- 46. Болезни нервной системы Эпилепсия Миопатия Лейкодистрофии Торсионная дистония Атаксия Фридрейха Болезнь Верднига-Гоффманна
- 47. Эпилепсия ЭПИЛЕПСИЯ – хроническое заболевание головного мозга, характеризующееся повторными приступами, которые возникают в результате чрезмерной нейронной
- 48. Эпилепсия
- 49. Миопатия Миопатия (от mio и греч. páthos - страдание, болезнь) – прогрессирующие мышечные дистрофии; относятся к
- 50. Лейкодистрофии Метахроматическая лейкодистрофия (лейкодистрофия Шольца-Гринфилда) относится к болезням накопления липидов. Основным патогенетическим механизмом дебюта и эволюции
- 51. Торсионная дистония ТОРСИОННАЯ ДИСТОНИЯ – наследственное заболевание нервной системы, характеризующееся нарушением мышечного тонуса и своеобразными двигательными
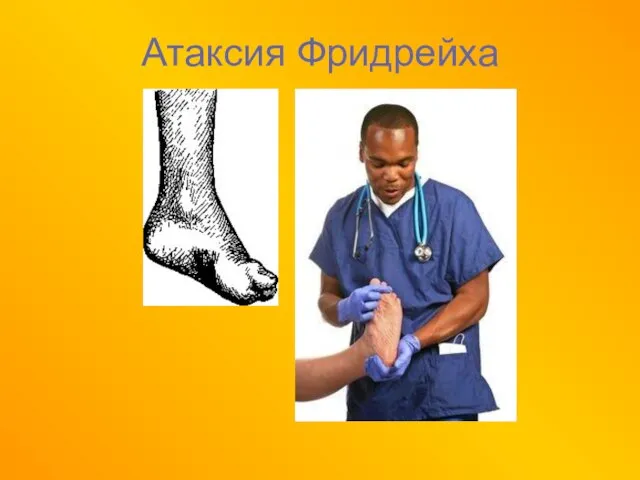
- 52. Атаксия Фридрейха Наследственное прогрессирующее заболевание нервной системы, характеризующееся нарушением координации, спастическими явлениями и изменениями скелета. В
- 53. Атаксия Фридрейха
- 54. Атаксия Фридрейха
- 55. Болезнь Вердинга-Гоффмана (амиотрофия наследственная спинальная) Аутосомно-рецессивное наследственное заболевание нервно-мышечной системы. Происходит дегенерация клеток передних рогов спинного
- 56. Болезнь Вердинга-Гоффмана (амиотрофия наследственная спинальная)
- 57. Поражения глаз Синдром Лоу Дегенерация роговицы Бюклерса Ретинобластома Хориоидеремия
- 58. Синдром Лоу Синдром Лоу – наследственная болезнь, характеризующаяся карликовостью, поражением глаз (катаракта и глаукома), умственной отсталостью,
- 59. Синдром Лоу
- 60. Дегенерация роговицы Бюклерса Дегенерация роговицы Бюклерса — наследственная болезнь глаз, проявляющаяся в детском возрасте и характеризующаяся
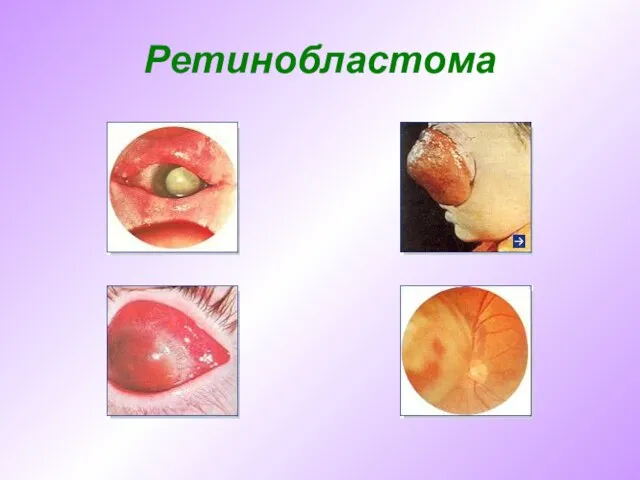
- 61. Ретинобластома Ретинобластома – злокачественная опухоль глаза, развивающаяся преимущественно в детском возрасте из тканей эмбрионального происхождения. Пик
- 62. Ретинобластома
- 63. Хориоидеремия Хориоидеремия – наследственная болезнь глаз, проявляющаяся понижением остроты зрения, концентрическим сужением полей зрения, гемералопией и
- 64. Болезнь Вильсона-Коновалова Болезнь Жильбера Болезнь Рандю-Вебера-Ослера Болезнь Виллебранда Глютеновая энтеропатия (целиакия) взрослых Болезни пищеварительной системы
- 65. Болезнь Вильсона-Коновалова Аутосомно-рецессивное наследственное заболевание, связанное с нарушением функции печени и обмена меди. Этиология и патогенез.
- 66. Болезнь Жильбера Болезнь Жильбера (пигментный гепатоз, ювенильная перемежающаяся желтуха) относится к наследственным доброкачественным хроническим заболеваниям, передающимся
- 67. Болезнь Рондю-Вебера-Ослера Болезнь обусловлена наследственной, передаваемой по аутосомно-доминантному типу неполноценностью сосудистого эндотелия, что приводит к ранимости
- 68. Болезнь Виллебранда Наследственная комбинированная геморрагическая гемостазиопатия, которая встречается у мужчин и женщин. Наследуется по аутосомно-доминантному типу.
- 70. Скачать презентацию
Слайд 2Словарик
Хромосомные болезни – наследственные заболевания, обусловленные изменением числа или структуры хромосом. Частота
Словарик
Хромосомные болезни – наследственные заболевания, обусловленные изменением числа или структуры хромосом. Частота

Слайд 3Моносомия - отсутствие в хромосомном наборе диплоидного организма одной хромосомы. Клетку или
Моносомия - отсутствие в хромосомном наборе диплоидного организма одной хромосомы. Клетку или

Слайд 4Типы наследственности
1. Аутосомно-доминантный тип наследования:
а. При достаточном числе потомков признак обнаруживается
Типы наследственности
1. Аутосомно-доминантный тип наследования:
а. При достаточном числе потомков признак обнаруживается

Слайд 5Наследственные заболевания:
Хромосомные болезни
Болезни обмена веществ
Нарушения иммунитета
Болезни с преимущественным поражением эндокринной системы
Болезни крови
Нарушение
Наследственные заболевания:
Хромосомные болезни
Болезни обмена веществ
Нарушения иммунитета
Болезни с преимущественным поражением эндокринной системы
Болезни крови
Нарушение
Слайд 6Хромосомные болезни:
Синдром Патау
Синдром Дауна
Синдром Эдвардса
Синдром Шершевского-Тернера
Синдром «кошачьего крика»
Синдром Клайнфельтера
Синдром дубль-Y
Трисомия
Хромосомные болезни:
Синдром Патау
Синдром Дауна
Синдром Эдвардса
Синдром Шершевского-Тернера
Синдром «кошачьего крика»
Синдром Клайнфельтера
Синдром дубль-Y
Трисомия
Слайд 7Синдром Шершевского-Тернера
Известно, что пол женщины и мужчины определяется наличием двух половых хромосом:
Синдром Шершевского-Тернера
Известно, что пол женщины и мужчины определяется наличием двух половых хромосом:

Слайд 8Синдром Шершевского-Тернера
Синдром Шершевского-Тернера
Слайд 9Синдром Патау
(синдром 13-трисомии)
Встречается примерно в одном случае на 25 тыс. живорожденных
Синдром Патау
(синдром 13-трисомии)
Встречается примерно в одном случае на 25 тыс. живорожденных

Слайд 10Синдром Патау
Синдром Патау
Слайд 11Синдром Эдвардса
(синдром 18-трисомии)
Встречается в одном случае на 6600 живорожденных, почти 80% пораженных
Синдром Эдвардса
(синдром 18-трисомии)
Встречается в одном случае на 6600 живорожденных, почти 80% пораженных

Слайд 12Синдром Эдвардса
Синдром Эдвардса

Слайд 13Синдром Дауна
(синдром 21-трисомии)
Встречается в одном случае на 7—10 тыс. живорожденных детей обоих
Синдром Дауна
(синдром 21-трисомии)
Встречается в одном случае на 7—10 тыс. живорожденных детей обоих

Слайд 14Синдром Дауна
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три
Синдром Дауна
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три
Слайд 15А теперь немного о мало известных хромосомных заболеваниях
человека...
А теперь немного о мало известных хромосомных заболеваниях
человека...

Слайд 16Синдром «кошачьего крика»
Синдром кошачьего крика (5р-) обусловлен делецией короткого плеча 5-й хромосомы.
Синдром «кошачьего крика»
Синдром кошачьего крика (5р-) обусловлен делецией короткого плеча 5-й хромосомы.

Слайд 17Синдром «кошачьего крика»
Синдром «кошачьего крика»

Слайд 18Синдром Клайнфельтера
Синдром Клайнфельтера включает случаи полисомии по половым хромосомам, при которых имеется
Синдром Клайнфельтера
Синдром Клайнфельтера включает случаи полисомии по половым хромосомам, при которых имеется

Слайд 19Синдром Клайнфельтера
Синдром Клайнфельтера

Слайд 20Синдром дубль-Y
Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г.
Синдром дубль-Y
Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г.

Слайд 21Трисомия Х
Частота трисомии-Х составляет среди новорожденных девочек и женщин 1:1000, среди умственно
Трисомия Х
Частота трисомии-Х составляет среди новорожденных девочек и женщин 1:1000, среди умственно

Слайд 22Болезни обмена веществ:
Гомоцистинурия
Гистидинемия
Нарушения обмена триптофана
Глюкоцереброзидозы
Финелкетонурия
Болезни обмена веществ:
Гомоцистинурия
Гистидинемия
Нарушения обмена триптофана
Глюкоцереброзидозы
Финелкетонурия
Слайд 23Глюкоцереброзидозы
Болезнь Гоше – это наиболее часто встречающаяся наследственная болезнь нарушения накопления, при
Глюкоцереброзидозы
Болезнь Гоше – это наиболее часто встречающаяся наследственная болезнь нарушения накопления, при

Слайд 24Фенилкетонурия
Фенилкетонурия (ФКУ) - тяжелое наследственное заболевание, наступающее вследствие врожденного дефекта фермента, отвечающего
Фенилкетонурия
Фенилкетонурия (ФКУ) - тяжелое наследственное заболевание, наступающее вследствие врожденного дефекта фермента, отвечающего

Слайд 25Гомоцистинурия
Заболевание наследуется по аутосомно-рецессивному типу. Частота гомоцистинурии составляет 1:200 000 новорожденных. В
Гомоцистинурия
Заболевание наследуется по аутосомно-рецессивному типу. Частота гомоцистинурии составляет 1:200 000 новорожденных. В

Слайд 26Гистидинемия
Заболевание возникает в результате отсутствия или недостаточности активности фермента гистидазы. Наследуется аутосомно-рецессивно.
Гистидинемия
Заболевание возникает в результате отсутствия или недостаточности активности фермента гистидазы. Наследуется аутосомно-рецессивно.

Слайд 27Нарушения обмена триптофана
Болезнь Гартнепа. Аутосомно-рецессивный тип наследования. При данном заболевании наблюдается генетическое
Нарушения обмена триптофана
Болезнь Гартнепа. Аутосомно-рецессивный тип наследования. При данном заболевании наблюдается генетическое

Слайд 28Нарушения иммунитета
Агаммаглобулинемия
Нарушения иммунитета
Агаммаглобулинемия

Слайд 29Агаммаглобулинемия
АГАММАГЛОБУЛИНЕМИЯ (греч.отриц. приставка а- + гамма-глобулин + греч. haima кровь; син.: пангипогамма-глобулинемия,
Агаммаглобулинемия
АГАММАГЛОБУЛИНЕМИЯ (греч.отриц. приставка а- + гамма-глобулин + греч. haima кровь; син.: пангипогамма-глобулинемия,

Слайд 30Болезни с преимущественным поражением эндокринной системы
Синдром Бёрьесона
Муковисцидоз
Кретинизм
Лепречаунизм
Болезни с преимущественным поражением эндокринной системы
Синдром Бёрьесона
Муковисцидоз
Кретинизм
Лепречаунизм

Слайд 31Синдром Бёрьесона
Бёрьесона-Форсмана-Лемана синдром – наследственная форма слабоумия с ожирением. Психика характеризуется умственным
Синдром Бёрьесона
Бёрьесона-Форсмана-Лемана синдром – наследственная форма слабоумия с ожирением. Психика характеризуется умственным

Слайд 32Муковисцидоз
Муковисцидоз - (от латинского mucus - слизь, viscidus - вязкий) самое распространенное
Муковисцидоз
Муковисцидоз - (от латинского mucus - слизь, viscidus - вязкий) самое распространенное

Слайд 33Кретинизм
КРЕТИНИЗМ - (от франц. cretin - слабоумный, кретин), эндокринное заболевание - недостаточная
Кретинизм
КРЕТИНИЗМ - (от франц. cretin - слабоумный, кретин), эндокринное заболевание - недостаточная

Слайд 34Кретинизм
На одном из портретов великого Веласкеса изображен Франциско Лескано, неряшливо одетый подросток
Кретинизм
На одном из портретов великого Веласкеса изображен Франциско Лескано, неряшливо одетый подросток

Слайд 35Лепречаунизм
Лепречаунизм (leprechaunismus; ирланд. leprechaun гном; син. Донохью синдром) - наследственная болезнь женщин,
Лепречаунизм
Лепречаунизм (leprechaunismus; ирланд. leprechaun гном; син. Донохью синдром) - наследственная болезнь женщин,

Слайд 36Болезни крови
Гемоглобинопатии
Серповидноклеточная анемия
Талассемия
Гемофилия
Тромбофилии
Болезни крови
Гемоглобинопатии
Серповидноклеточная анемия
Талассемия
Гемофилия
Тромбофилии
Слайд 37Гемоглобинопатии
Гемоглобинопатии (от гемоглобин и греч. páthos - страдание, болезнь), гемоглобинозы – состояния,
Гемоглобинопатии
Гемоглобинопатии (от гемоглобин и греч. páthos - страдание, болезнь), гемоглобинозы – состояния,

Слайд 38Серповидноклеточная анемия
Серповидноклеточная анемия (HbS) связана с наличием в эритроцитах патологического гемоглобина S.
Серповидноклеточная анемия
Серповидноклеточная анемия (HbS) связана с наличием в эритроцитах патологического гемоглобина S.

Слайд 39Талассемия
Талассемия - заболевание, распространённое в средиземноморских странах. Характеризуется значительным повышением содержания HbF
Талассемия
Талассемия - заболевание, распространённое в средиземноморских странах. Характеризуется значительным повышением содержания HbF

Слайд 40Гемофилия
Гемофилия – Рецессивно наследуемое заболевание, сцепленное с Х- хромосомой. Гемофилия А -
Гемофилия
Гемофилия – Рецессивно наследуемое заболевание, сцепленное с Х- хромосомой. Гемофилия А -

Слайд 41Гемофилия
Гемофилия
Слайд 42Тромбофилии
ТРОМБОФИЛИИ ГЕМАТОГЕННЫЕ – состояния, характеризующиеся наклонностью к развитию рецидивирующих тромбозов кровеносных сосудов
Тромбофилии
ТРОМБОФИЛИИ ГЕМАТОГЕННЫЕ – состояния, характеризующиеся наклонностью к развитию рецидивирующих тромбозов кровеносных сосудов

Слайд 43Нарушение функций почек
Фосфат-диабет
Наследственный нефрит
Нарушение функций почек
Фосфат-диабет
Наследственный нефрит

Слайд 44Фосфат-диабет
ФОСФАТ-ДИАБЕТ – доминантно сцепленное с Х-хромосомой заболевание с глубокими нарушениями фосфорно-кальциевого обмена,
Фосфат-диабет
ФОСФАТ-ДИАБЕТ – доминантно сцепленное с Х-хромосомой заболевание с глубокими нарушениями фосфорно-кальциевого обмена,

Слайд 45Наследственный нефрит
НАСЛЕДСТВЕННЫЙ НЕФРИТ. Этиология, патогенез не изучены. Предполагается, что заболевание связано с
Наследственный нефрит
НАСЛЕДСТВЕННЫЙ НЕФРИТ. Этиология, патогенез не изучены. Предполагается, что заболевание связано с

Слайд 46Болезни нервной системы
Эпилепсия
Миопатия
Лейкодистрофии
Торсионная дистония
Атаксия Фридрейха
Болезнь Верднига-Гоффманна
Болезни нервной системы
Эпилепсия
Миопатия
Лейкодистрофии
Торсионная дистония
Атаксия Фридрейха
Болезнь Верднига-Гоффманна

Слайд 47Эпилепсия
ЭПИЛЕПСИЯ – хроническое заболевание головного мозга, характеризующееся повторными приступами, которые возникают в
Эпилепсия
ЭПИЛЕПСИЯ – хроническое заболевание головного мозга, характеризующееся повторными приступами, которые возникают в

Слайд 48Эпилепсия
Эпилепсия

Слайд 49Миопатия
Миопатия (от mio и греч. páthos - страдание, болезнь) – прогрессирующие мышечные
Миопатия
Миопатия (от mio и греч. páthos - страдание, болезнь) – прогрессирующие мышечные

Слайд 50Лейкодистрофии
Метахроматическая лейкодистрофия (лейкодистрофия Шольца-Гринфилда) относится к болезням накопления липидов. Основным патогенетическим механизмом
Лейкодистрофии
Метахроматическая лейкодистрофия (лейкодистрофия Шольца-Гринфилда) относится к болезням накопления липидов. Основным патогенетическим механизмом

Слайд 51Торсионная дистония
ТОРСИОННАЯ ДИСТОНИЯ – наследственное заболевание нервной системы, характеризующееся нарушением мышечного тонуса
Торсионная дистония
ТОРСИОННАЯ ДИСТОНИЯ – наследственное заболевание нервной системы, характеризующееся нарушением мышечного тонуса

Слайд 52Атаксия Фридрейха
Наследственное прогрессирующее заболевание нервной системы, характеризующееся нарушением координации, спастическими явлениями и
Атаксия Фридрейха
Наследственное прогрессирующее заболевание нервной системы, характеризующееся нарушением координации, спастическими явлениями и

Слайд 53Атаксия Фридрейха
Атаксия Фридрейха
Слайд 54Атаксия Фридрейха
Атаксия Фридрейха

Слайд 55Болезнь Вердинга-Гоффмана
(амиотрофия наследственная спинальная)
Аутосомно-рецессивное наследственное заболевание нервно-мышечной системы. Происходит дегенерация клеток передних
Болезнь Вердинга-Гоффмана
(амиотрофия наследственная спинальная)
Аутосомно-рецессивное наследственное заболевание нервно-мышечной системы. Происходит дегенерация клеток передних

Слайд 56Болезнь Вердинга-Гоффмана
(амиотрофия наследственная спинальная)
Болезнь Вердинга-Гоффмана
(амиотрофия наследственная спинальная)

Слайд 57Поражения глаз
Синдром Лоу
Дегенерация роговицы Бюклерса
Ретинобластома
Хориоидеремия
Поражения глаз
Синдром Лоу
Дегенерация роговицы Бюклерса
Ретинобластома
Хориоидеремия
Слайд 58Синдром Лоу
Синдром Лоу – наследственная болезнь, характеризующаяся карликовостью, поражением глаз (катаракта и
Синдром Лоу
Синдром Лоу – наследственная болезнь, характеризующаяся карликовостью, поражением глаз (катаракта и

Слайд 59Синдром Лоу
Синдром Лоу

Слайд 60Дегенерация роговицы Бюклерса
Дегенерация роговицы Бюклерса — наследственная болезнь глаз, проявляющаяся в детском
Дегенерация роговицы Бюклерса
Дегенерация роговицы Бюклерса — наследственная болезнь глаз, проявляющаяся в детском

Слайд 61Ретинобластома
Ретинобластома – злокачественная опухоль глаза, развивающаяся преимущественно в детском возрасте из тканей
Ретинобластома
Ретинобластома – злокачественная опухоль глаза, развивающаяся преимущественно в детском возрасте из тканей

Слайд 62Ретинобластома
Ретинобластома
Слайд 63Хориоидеремия
Хориоидеремия – наследственная болезнь глаз, проявляющаяся понижением остроты зрения, концентрическим сужением
Хориоидеремия
Хориоидеремия – наследственная болезнь глаз, проявляющаяся понижением остроты зрения, концентрическим сужением

Слайд 64Болезнь Вильсона-Коновалова
Болезнь Жильбера
Болезнь Рандю-Вебера-Ослера
Болезнь Виллебранда
Глютеновая энтеропатия (целиакия) взрослых
Болезни
пищеварительной
системы
Болезнь Вильсона-Коновалова
Болезнь Жильбера
Болезнь Рандю-Вебера-Ослера
Болезнь Виллебранда
Глютеновая энтеропатия (целиакия) взрослых
Болезни
пищеварительной
системы
Слайд 65Болезнь Вильсона-Коновалова
Аутосомно-рецессивное наследственное заболевание, связанное с нарушением функции печени и обмена меди.
Болезнь Вильсона-Коновалова
Аутосомно-рецессивное наследственное заболевание, связанное с нарушением функции печени и обмена меди.

Слайд 66Болезнь Жильбера
Болезнь Жильбера (пигментный гепатоз, ювенильная перемежающаяся желтуха) относится к наследственным доброкачественным
Болезнь Жильбера
Болезнь Жильбера (пигментный гепатоз, ювенильная перемежающаяся желтуха) относится к наследственным доброкачественным

Слайд 67Болезнь Рондю-Вебера-Ослера
Болезнь обусловлена наследственной, передаваемой по аутосомно-доминантному типу неполноценностью сосудистого эндотелия, что
Болезнь Рондю-Вебера-Ослера
Болезнь обусловлена наследственной, передаваемой по аутосомно-доминантному типу неполноценностью сосудистого эндотелия, что

Слайд 68Болезнь Виллебранда
Наследственная комбинированная геморрагическая гемостазиопатия, которая встречается у мужчин и женщин. Наследуется
Болезнь Виллебранда
Наследственная комбинированная геморрагическая гемостазиопатия, которая встречается у мужчин и женщин. Наследуется

 Как подготовить конспект урока
Как подготовить конспект урока ОГБОУНПОПрофессиональное училище №12.
ОГБОУНПОПрофессиональное училище №12. Многожанровость музыкального искусства
Многожанровость музыкального искусства Организация трансграничного электронного документооборота на основе доверенной третьей стороны
Организация трансграничного электронного документооборота на основе доверенной третьей стороны Антинея
Антинея Мониторинг гражданского обществав Российской Федерации
Мониторинг гражданского обществав Российской Федерации Арктические тундры
Арктические тундры Презентация на тему Знаки препинания при вводных словах и предложениях
Презентация на тему Знаки препинания при вводных словах и предложениях История полиции в Новониколаевске
История полиции в Новониколаевске Физики и деньги
Физики и деньги Кроссплатформенное СПО как альтернатива проприетарному
Кроссплатформенное СПО как альтернатива проприетарному Пасхальный калейдоскоп
Пасхальный калейдоскоп Презентация
Презентация Правописание частицы НЕ с разным частями речи
Правописание частицы НЕ с разным частями речи Мощные производители из наиболее развитых провинций - Цзянсу
Мощные производители из наиболее развитых провинций - Цзянсу Горы России 4 КЛАСС
Горы России 4 КЛАСС Советская музыка
Советская музыка Школьный этап конкурса «Учитель года -2010»
Школьный этап конкурса «Учитель года -2010» Презентация на тему Скорость сближения и удаления
Презентация на тему Скорость сближения и удаления Цели внедрения системы бюджетирования
Цели внедрения системы бюджетирования Презентация на тему Отряд Подёнки
Презентация на тему Отряд Подёнки Повторение темы «Наречие».
Повторение темы «Наречие». Миф о Персее. Созвездия Цефея, Кассиопеи, Персея, Андромеды и Пегаса.
Миф о Персее. Созвездия Цефея, Кассиопеи, Персея, Андромеды и Пегаса. Звуковые колебания
Звуковые колебания Не умри, и мы отстанем
Не умри, и мы отстанем Презентация на тему Международные отношения в 1920-30-е годы
Презентация на тему Международные отношения в 1920-30-е годы  Художественный стиль:
Художественный стиль: Программа курса по выбору «Основы уголовного права».
Программа курса по выбору «Основы уголовного права».