- Организация наследственного аппарата в клетках человека

Содержание
- 2. Исторические этапы изучения организации и функционирования наследственного аппарата 1865 г. Ф.Гальтон - «Наследование таланта и характера».
- 3. 1910-1925 гг. Т.Морган – положения хромосомной теории наследственности. 1926 г. Х.Дж.Мёллер – мутагенное действие рентгеновских лучей.
- 4. Наследование – это процесс передачи генетической информации в ряду поколений. Наследуемые признаки могут быть качественными (моногенными)
- 5. Грегор Мендель – основатель генетики Первый закон Менделя Закон единообразия гибридов первого поколения, или закон доминирования.
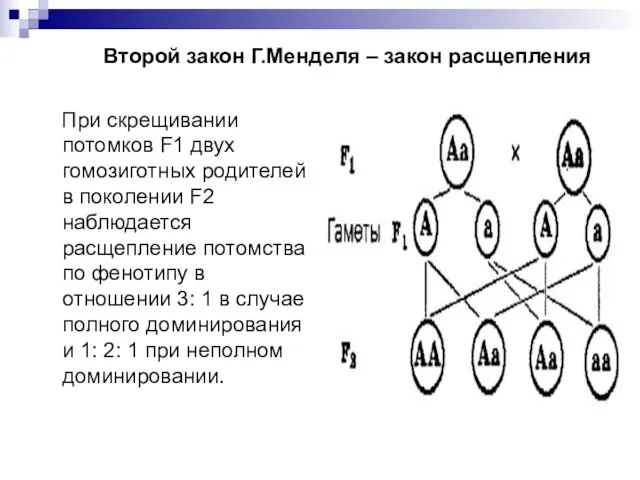
- 6. Второй закон Г.Менделя – закон расщепления При скрещивании потомков F1 двух гомозиготных родителей в поколении F2
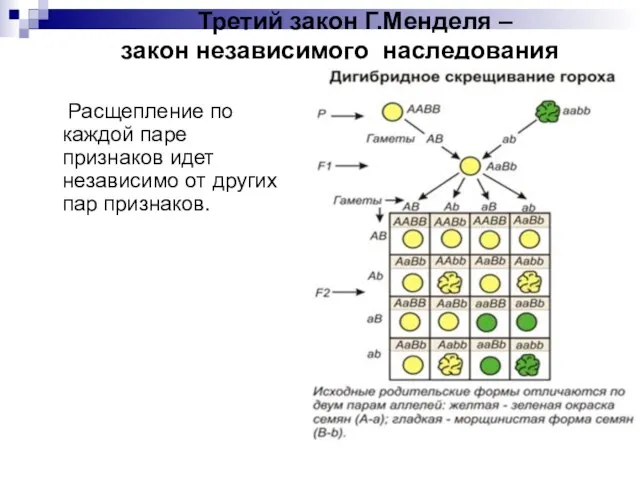
- 7. Третий закон Г.Менделя – закон независимого наследования Расщепление по каждой паре признаков идет независимо от других
- 8. Анализирующее скрещивание Чтобы выяснить генотип гибрида второго поколения за одно скрещивание, необходимо произвести возвратное (анализирующее) скрещивание
- 9. В кариотипе человека содержится 44 аутосомы и 2 половых хромосомы – Х и Y. Женский пол
- 10. Анализируя механизмы сцепленного наследования Т. Морган и его сотрудники сформулировали положения хромосомной теории. Основные положения хромосомной
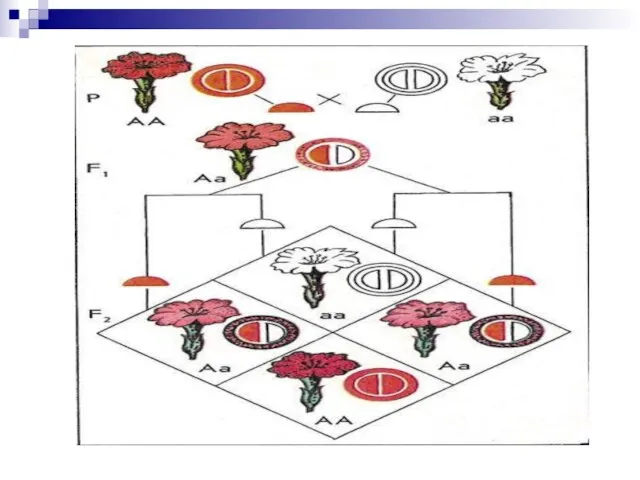
- 11. Взаимодействия аллельных генов Типы доминирования: Полное доминирование. Неполное доминирование. Отмечается в случаях, когда фенотип гетерозигот Аа
- 13. Кодоминирование. Это такой тип взаимодействия аллельных генов, при котором каждый из аллелей проявляет свое действие. В
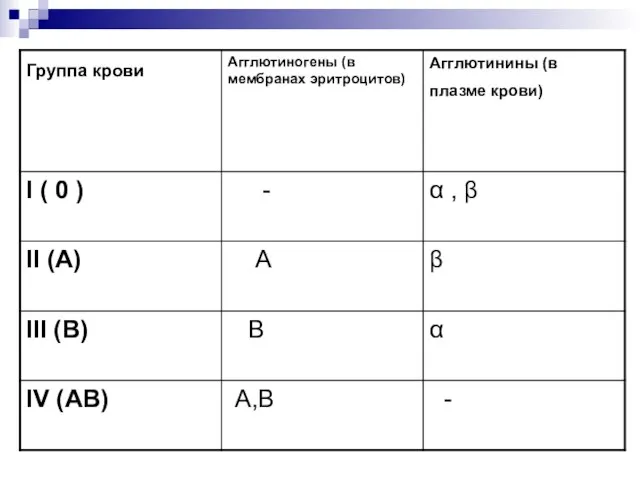
- 14. Группы крови - это генетически наследуемые признаки, не изменяющиеся в течение жизни при естественных условиях. Группа
- 16. Наследование групп крови человека системы АВО
- 17. Переливание крови - это введение определенного количества донорской крови в кровь реципиента. Человек, дающий кровь для
- 19. Несовместимость крови наблюдается, если эритроциты одной крови несут агглютиногены (А или В), а в плазме другой
- 20. Резус-фактор белок на мембране эритроцитов. Присутствует у 85% людей - резус-положительных. Остальные 15% - резус-отрицательны. Наследование:
- 21. Взаимодействие неаллельных генов: Комплементарным называется взаимодействие, при котором действие генов из одной пары дополняется действием генов
- 22. Полимерия – взаимодействие неаллельных множественных генов, однозначно влияющих на развитие одного и того же признака; степень
- 23. У человека может наблюдаться предрасположенность к различным заболеваниям: гипертонической болезни, ожирению, сахарному диабету, шизофрении и др.
- 24. Эпистаз – взаимодействие неаллельных генов, при котором один из них подавляется другим. Подавляющий ген называется эпистатичным,
- 25. Доминантный эпистаз. При доминантном эпистазе действие доминантных генов из одной пары подавляет работу также доминантных генов
- 26. Под «эффектом положения» понимают взаимное влияние генов разных аллелей, занимающих близлежащие локусы в одной хромосоме. Оно
- 27. Виды изменчивости: 1. Наследственная (генотипическая) изменчивость связана с изменением самого генетического материала. 2. Ненаследственная (фенотипическая, модификационная)
- 28. Комбинативная изменчивость Связана с новым сочетанием неизменных генов родителей в генотипах потомства. Факторы комбинативной изменчивости. 1.
- 29. Генетика пола Ни один природный феномен не привлекает к себе такого внимания и не содержит столько
- 30. Основные механизмы определения пола Прогамное – пол определяется до оплодотворения. Характерно для особей, размножающихся партеногенетически. Так,
- 31. У некоторых видов в ходе обычного онтогенеза при определенных условиях происходит естественное переопределение пола. В Тихом
- 32. Дифференцировка пола в процессе развития Процесс первичной дифференцировки пола связан с периодом эмбрионального развития. Формирование закладок
- 33. Соотношение полов у человека Теоретически соотношение полов у человека должно быть 1:1 (50%:50%), т.к. встреча яйцеклетки
- 36. Мутационная изменчивость Мутации - это скачкообразные изменения генетического материала под влиянием факторов внешней или внутренней среды.
- 37. Мутагенные факторы: К физическим мутагенам относятся различные виды излучений (преимущественно ионизирующих), высокая температура, УФ- лучи, СВЧ
- 39. Классификация мутаций наследственного аппарата Спонтанные- возникают под влиянием неизвестного природного фактора, чаще всего как результат ошибок
- 40. 2)По локализации: Генеративные или гаметические происходят в процессе образования половых клеток (нарушения мейоза) или в клетках,
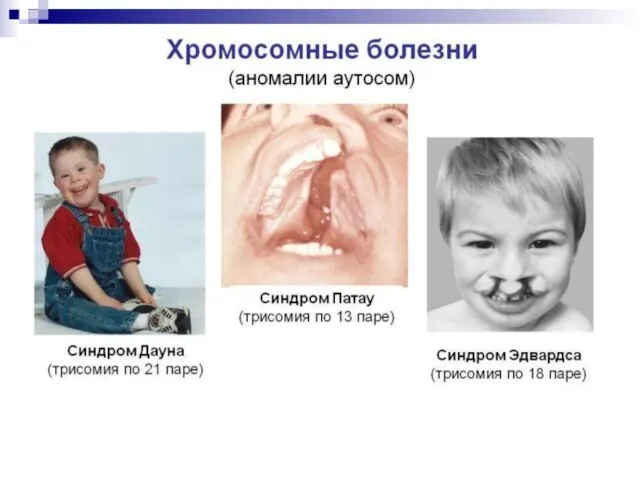
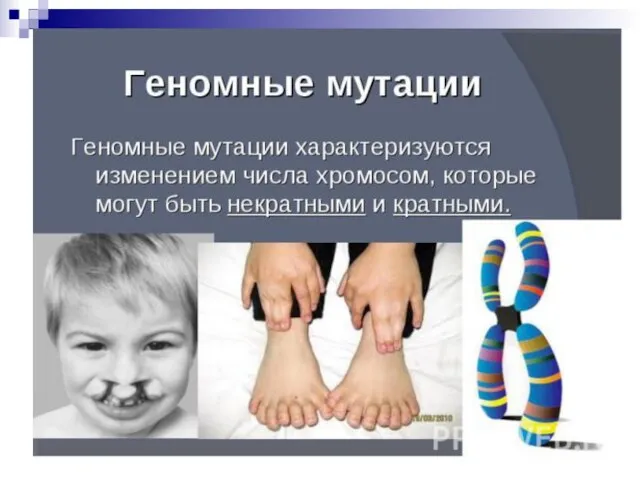
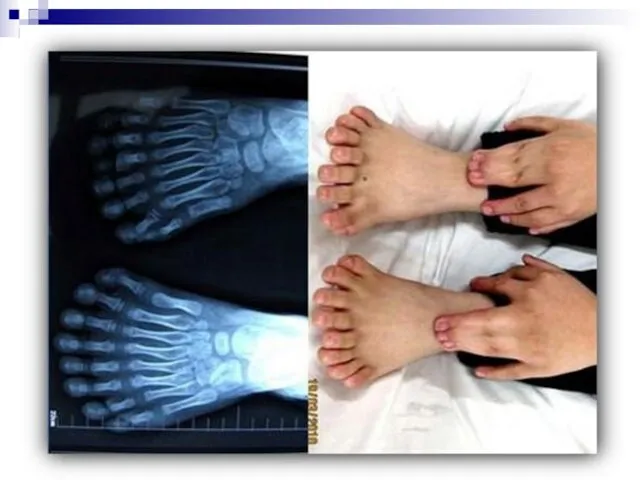
- 44. Все хромосомные болезни можно разделить на 2 группы: Геномные, связанные с аномалиями числа хромосом; Хромосомные, связанные
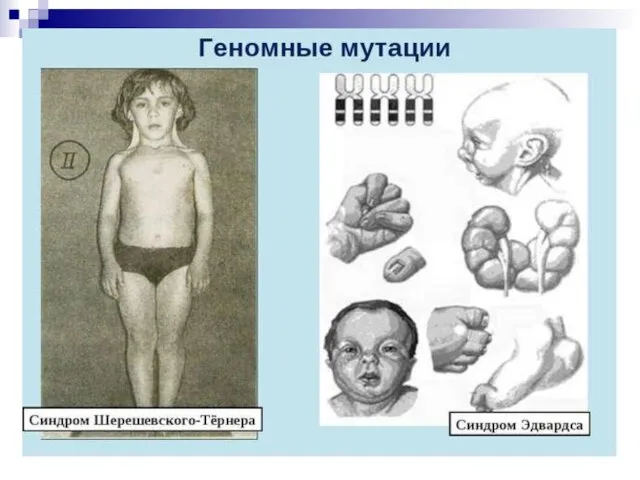
- 46. 4) По уровню организации наследственного аппарата: Геномные мутации обусловлены изменением числа хромосом. Причины: а)нерасхождения хромосом, когда
- 49. Гетероплоидия, или анеуплоидия - некратное гаплоидному уменьшение или увеличение числа хромосом (2n+1, 2n+2, 2n-1 и т.д.).
- 50. Гетероплоидия —изменение числа хромосом, не кратное гаплоидному набору. При этом набор хромосом в клетке может быть
- 52. Новорожденный с синдромом Дауна отличается характерным внешним видом: округлый череп со скошенным затылком, косой разрез глаз,
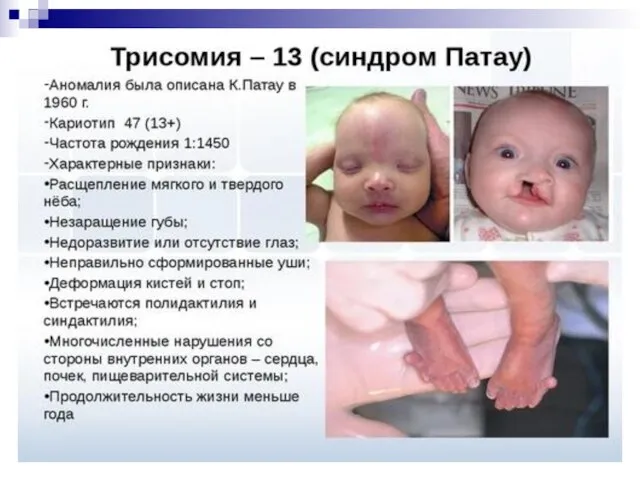
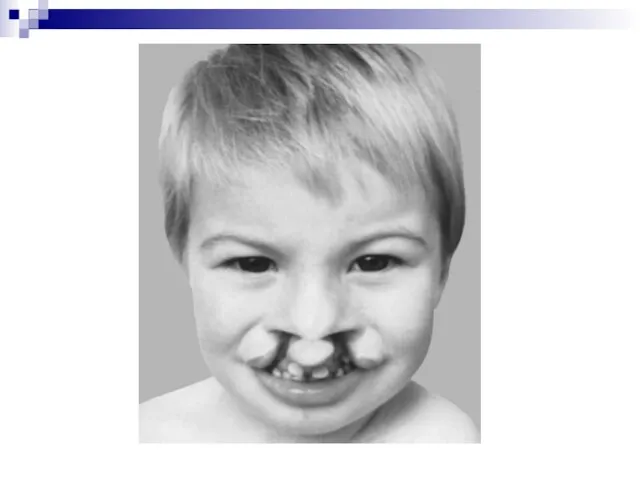
- 53. синдром Патау (трисомия по 13 паре аутосом), характеризующийся отсутствием шеи, различными уродствами на лице, неспособностью к
- 58. Аномалии половых хромосом 47, ХХХ – трисомия Х Частота 1:800, 1:1000. Фенотип женский. Недоразвитие яичников в
- 61. 47, ХХY; 48, XXXY и др. – синдром Клайнфельтера. Частота – 1:400, 1:500. Фенотип мужской. Телосложение
- 65. Помимо изменений в числе хромосом, известны и случаи изменений в структуре хромосом, приводящие к умственной неполноценности.
- 70. Хромосомная патология является одним из основных факторов формирования множественных пороков развития и составляют 30% от общего
- 71. Хромосомные мутации (абберации) обусловлены изменением структуры хромосом. К внутрихромосомным мутациям относятся перестройки внутри одной хромосомы. а)
- 74. ХРОМОСОМНАЯ ТЕРМИНОЛОГИЯ 1. В самом начале указывается общее число хромосом: 46,45,47. 2.Состав половых хромосом: 46.ХХ -нормальный
- 75. Генные (точковые) мутации связанны с изменением структуры гена (молекулы ДНК), могут затрагивать как структурные гены, так
- 76. Генные мутации у человека являются причинами многих наследственных моногенных заболеваний. При изучении белковых продуктов мутантных генов
- 77. Фенотипически генные мутации проявляются как наследственные болезни обмена веществ - ферментопатии. Вещества, накапливающиеся в результате отсутствия
- 78. Наиболее часто встречающимися болезнями, связанными с нарушением аминокислотного обмена являются: фенилкетонурия и альбинизм. В норме аминокислота
- 80. Альбинизм встречается в разных популяциях с разной частотой - от 1:5000 до 1:25000. Он наследуется по
- 82. Алкаптонурия встречается довольно редко (3-5:1000000). Наследуется по аутосомно-рецессивному типу. Алкаптонурия является следствием генетического дефекта оксидазы, катализирующей
- 84. Наиболее частыми наследственными дефектами нарушения обмена углеводов являются галактоземия и мукополисахаридозы. Галактоземия встречается с частотой примерно
- 86. Наследственные дефекты обмена липидов : Эти болезни встречаются редко (1:300000). Высокая частота болезни Тея-Сакса наблюдается только
- 88. Гиперлипопротеинемии обусловлены нарушением обмена липидов плазмы крови вследствие дефектов ферментов или клеточных рецепторов. Липиды плазмы крови
- 90. Наследственные дефекты обмена пуринов и пиримидинов Синдром Леша-Нихана обусловлен недостаточностью фермента гипоксантин-фосфорибозилтрансферазы (ГФРТ), который катализирует присоединение
- 92. Примером нарушения минерального обмена может служить расстройство обмена меди. Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия) обусловлена генной мутацией,
- 95. Наследственные заболевания , вызванные нарушением развития органов и тканей. Муковисцидоз (кистофиброз поджелудочной железы) обусловлен генной мутацией
- 99. Ахондроплазия (хондродистрофия) обусловлена генной мутацией, вызывающей отклонения в активности некоторых ферментов (5- нуклеотидазы, глюкозо-6-фосфатазы). Тип наследования
- 101. Миодистрофия Дюшенна (МД) - тяжелое наследственное заболевание обусловленное мутацией гена дистрофина, что приводит к дегенерации мышечных
- 103. Синдром фрагильной (ломкой) Х-хромосомы - тяжелое наследственное заболевание. Тип наследования – Х-сцепленный доминантный с неполной пенетрантностью
- 105. Наследственные заболевания нарушения свертывающей системы крови. Гемофилия А - тяжелое наследственное заболевание, обусловленное дефектом VIII фактором
- 106. Гемоглобинопатии - заболевания, связанные с нарушением структуры молекулы гемоглобина. Нормальный гемоглобин человека (HbA) состоит из двух
- 121. Повторим лекцию
- 122. Виды наследования Моногенное - тип наследования, при котором признак определяется ОДНИМ геном. Полигенное - тип наследования,
- 124. Моногенное наследование — наследование одного признака. • аутомосный доминантный; • аутосомный рецессивный; • Х-сцепленный доминантный; •
- 125. Доминантное наследование Доминантное наследование — когда признак кодируется доминантным геном. Ген считается доминантным, если кодируемый им
- 126. Примером доминантного наследования является наследование заболевания Хорея Гентингтона. Хорея Гентингтона — дегенеративное заболевание нервных клеток в
- 129. Ещё одним примером доминантного моногенного наследования может служить брахидактилия (короткопалость). Анализ семейных форм проявления данного признака
- 131. Рецессивное наследование. Рецессивным ген считается, если признак, который он кодирует, не проявляется в присутствии противоположного гена.
- 132. Особенности рецессивного наследования: 1. Признак проявляется только у рецессивных гомозиготных особей, при генотипе (аа). 2. Возможно
- 139. К менделеевскому наследованию по доминантному-рецессивному типу относится и наследование резус-фактора крови. Ген, кодирующий резус-фактор Б, является
- 140. Резус-конфликт — это гуморальный иммунный ответ резус-отрицательной матери на эритроцитарные антигены резус-положительного плода, при котором образуются
- 141. Варианты возникновения резус-конфликта: • Если мать является резус-отрицательной, а отец — гомозиготным резус-положительным, то любой плод
- 142. В подавляющем большинстве случаев резус-конфликт может быть предупреждён путём внутримышечного введения резус-отрицательной матери специальных анти-Б антител)
- 143. Неполное доминирование — в этом случае гетерозигота занимает промежуточное положение между доминантной и рецессивной гомозиготой. Например,
- 144. Кодоминирование — в фенотипе гетерозиготы проявляются два признака. Примером может являться наследование четвёртой группы крови по
- 145. Наследование, сцепленное с полом. Гены могут находиться на половых хромосомах, в этом случае говорят, что они
- 146. Например, гемофилии — болезни, связанной с нарушением нормальной свёртываемости крови. При этих нарушениях у больного возникают
- 147. Самой известной носительницей гемофилии в истории была королева Виктория; по-видимому, эта мутация произошла в её генотипе
- 151. Среди других генов, сцепленных с полом, стоит упомянуть гены, связанные с цветовой слепотой и ихтиозом.
- 153. Встречаются и доминантные гены, сцепленные с Х-хромосомой Так, существует наследственная форма рахита, которая не поддаётся лечению
- 157. От наследования, сцепленного с полом, надо отличать наследование, ограниченное полом. В случае наследования, ограниченного полом, гены,
- 159. Митохондриальное наследование. Митохондриальная ДНК представляет из себя одну кольцевидно-замкнутую хромосому. Закономерности митохондриального наследования: • Болеют и
- 162. Полигенное наследование Полигенное наследование — наследование признаков, зависящих от нескольких генов.
- 163. Комплементарность такое взаимодействие генов, при котором 2 или более генов вызывают развитие признака. Например, у человека
- 164. Полимерия — несколько генов действуют на один признак одинаково. При этом при формировании признака не важно,
- 165. Плейотропия Плейотропия — влияние одного гена на появление нескольких признаков. Примером может служить аутосомно-доминантное заболевание из
- 170. Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который впервые представил описание 5-летней
- 171. Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30-40 годами и смерть наступает вследствие
- 173. Эпистаз подавление одним геном другого, неаллельного. Примером эпистаза может служить "бомбейский феномен". В Индии описаны семьи,
- 174. Ещё одним примером эпистаза может служить появление белых альбиносов в семье темнокожих. В данном случае рецессивный
- 176. Синдром Морриса — синдром нечувствительности к андрогенам (синдром тестикулярной феминизации) проявляется нарушениями полового развития, которые развиваются
- 177. Синдром тестикулярной феминизации (СТФ) впервые описан в 1817 году баварским врачом, он описывал, как при вскрытии
- 178. СТФ очень редок — по данным разных авторов, частота его составляет от 1:65 000 до 1:10
- 179. большинстве исследований пациентов с синдромом Морриса описана не только их физическая привлекательность, но и исключительная деловитость,
- 180. Великая «неженщины»: 7 особенностей, которые позволили утверждать, что у Жанны д'Арк был синдром Морриса. Жанна д'Арк
- 182. Скачать презентацию
Слайд 2 Исторические этапы изучения организации и функционирования наследственного аппарата
1865 г. Ф.Гальтон -
Исторические этапы изучения организации и функционирования наследственного аппарата
1865 г. Ф.Гальтон -

Слайд 31910-1925 гг. Т.Морган – положения хромосомной теории наследственности.
1926 г. Х.Дж.Мёллер –
1910-1925 гг. Т.Морган – положения хромосомной теории наследственности.
1926 г. Х.Дж.Мёллер –

Слайд 4 Наследование – это процесс передачи генетической информации в ряду поколений.
Наследуемые признаки
Наследование – это процесс передачи генетической информации в ряду поколений.
Наследуемые признаки

Слайд 5Грегор Мендель – основатель генетики
Первый закон Менделя
Закон единообразия гибридов первого поколения, или
Грегор Мендель – основатель генетики
Первый закон Менделя
Закон единообразия гибридов первого поколения, или

Слайд 6 Второй закон Г.Менделя – закон расщепления
При скрещивании потомков F1 двух
Второй закон Г.Менделя – закон расщепления
При скрещивании потомков F1 двух
Слайд 7 Третий закон Г.Менделя – закон независимого наследования
Расщепление по каждой паре
Третий закон Г.Менделя – закон независимого наследования
Расщепление по каждой паре
Слайд 8Анализирующее скрещивание
Чтобы выяснить генотип гибрида второго поколения за одно скрещивание, необходимо произвести
Анализирующее скрещивание
Чтобы выяснить генотип гибрида второго поколения за одно скрещивание, необходимо произвести

Слайд 9В кариотипе человека содержится 44 аутосомы и 2 половых хромосомы – Х
В кариотипе человека содержится 44 аутосомы и 2 половых хромосомы – Х

Слайд 10Анализируя механизмы сцепленного наследования Т. Морган и его сотрудники сформулировали положения хромосомной
Анализируя механизмы сцепленного наследования Т. Морган и его сотрудники сформулировали положения хромосомной

Слайд 11Взаимодействия аллельных генов
Типы доминирования:
Полное доминирование.
Неполное доминирование. Отмечается в
Взаимодействия аллельных генов
Типы доминирования:
Полное доминирование.
Неполное доминирование. Отмечается в

Слайд 13Кодоминирование. Это такой тип взаимодействия аллельных генов, при котором каждый из аллелей
Кодоминирование. Это такой тип взаимодействия аллельных генов, при котором каждый из аллелей

Слайд 14Группы крови - это генетически наследуемые признаки, не изменяющиеся в течение жизни
Группы крови - это генетически наследуемые признаки, не изменяющиеся в течение жизни

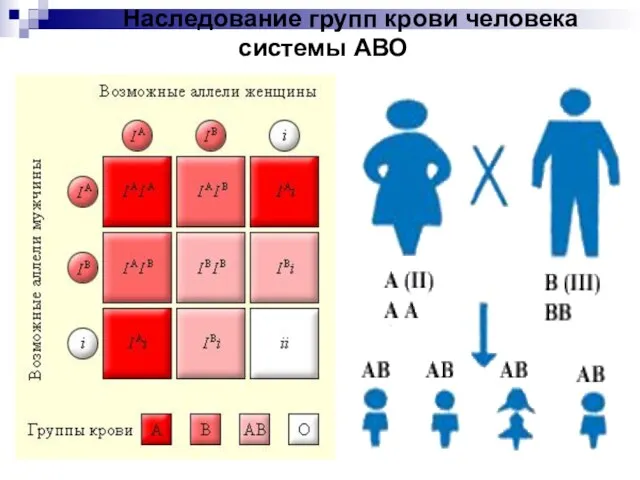
Слайд 16 Наследование групп крови человека системы АВО
Наследование групп крови человека системы АВО
Слайд 17Переливание крови - это введение определенного количества донорской крови в кровь реципиента.
Переливание крови - это введение определенного количества донорской крови в кровь реципиента.

Слайд 19Несовместимость крови наблюдается, если эритроциты одной крови несут агглютиногены (А или В),
Несовместимость крови наблюдается, если эритроциты одной крови несут агглютиногены (А или В),

Слайд 20Резус-фактор белок на мембране эритроцитов. Присутствует у 85% людей - резус-положительных. Остальные
Резус-фактор белок на мембране эритроцитов. Присутствует у 85% людей - резус-положительных. Остальные

Слайд 21 Взаимодействие неаллельных генов:
Комплементарным называется взаимодействие, при котором действие генов из
Взаимодействие неаллельных генов:
Комплементарным называется взаимодействие, при котором действие генов из

Слайд 22Полимерия – взаимодействие неаллельных множественных генов, однозначно влияющих на развитие одного и
Полимерия – взаимодействие неаллельных множественных генов, однозначно влияющих на развитие одного и

Слайд 23У человека может наблюдаться предрасположенность к различным заболеваниям: гипертонической болезни, ожирению, сахарному

Слайд 24Эпистаз – взаимодействие неаллельных генов, при котором один из них подавляется другим.

Слайд 25 Доминантный эпистаз. При доминантном эпистазе действие доминантных генов из одной пары подавляет
Доминантный эпистаз. При доминантном эпистазе действие доминантных генов из одной пары подавляет

Слайд 26Под «эффектом положения» понимают взаимное влияние генов разных аллелей, занимающих близлежащие локусы
Под «эффектом положения» понимают взаимное влияние генов разных аллелей, занимающих близлежащие локусы

Слайд 27Виды изменчивости:
1. Наследственная (генотипическая) изменчивость связана с изменением самого генетического материала.
2. Ненаследственная (фенотипическая,
1. Наследственная (генотипическая) изменчивость связана с изменением самого генетического материала.
2. Ненаследственная (фенотипическая,

Слайд 28Комбинативная изменчивость
Связана с новым сочетанием неизменных генов родителей в генотипах потомства. Факторы
Комбинативная изменчивость
Связана с новым сочетанием неизменных генов родителей в генотипах потомства. Факторы

Слайд 29Генетика пола
Ни один природный феномен не привлекает к себе такого внимания и
Генетика пола
Ни один природный феномен не привлекает к себе такого внимания и

Слайд 30Основные механизмы определения пола
Прогамное – пол определяется до оплодотворения. Характерно для особей,
Основные механизмы определения пола
Прогамное – пол определяется до оплодотворения. Характерно для особей,

Слайд 31У некоторых видов в ходе обычного онтогенеза при определенных условиях происходит естественное
У некоторых видов в ходе обычного онтогенеза при определенных условиях происходит естественное

Слайд 32Дифференцировка пола в процессе развития
Процесс первичной дифференцировки пола связан с периодом эмбрионального
Дифференцировка пола в процессе развития
Процесс первичной дифференцировки пола связан с периодом эмбрионального

Слайд 33Соотношение полов у человека
Теоретически соотношение полов у человека должно быть 1:1 (50%:50%),
Соотношение полов у человека
Теоретически соотношение полов у человека должно быть 1:1 (50%:50%),


Слайд 36Мутационная изменчивость
Мутации - это скачкообразные изменения генетического материала под влиянием факторов внешней
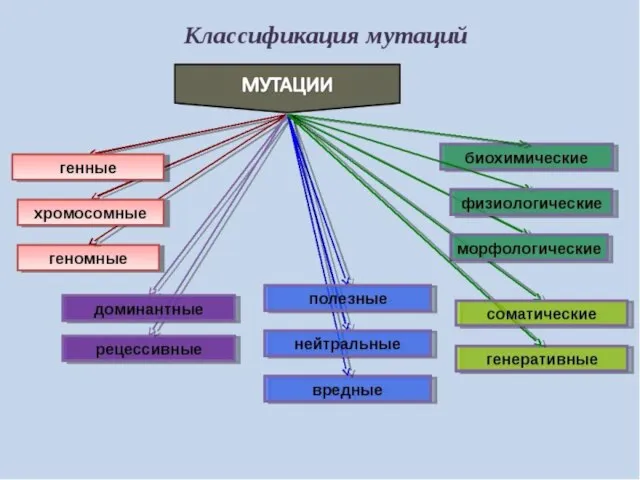
Мутационная изменчивость
Мутации - это скачкообразные изменения генетического материала под влиянием факторов внешней

Слайд 37Мутагенные факторы:
К физическим мутагенам относятся различные виды излучений (преимущественно ионизирующих), высокая температура,
Мутагенные факторы:
К физическим мутагенам относятся различные виды излучений (преимущественно ионизирующих), высокая температура,

Слайд 39 Классификация мутаций наследственного аппарата
Спонтанные- возникают под влиянием неизвестного природного фактора, чаще
Классификация мутаций наследственного аппарата
Спонтанные- возникают под влиянием неизвестного природного фактора, чаще

Слайд 402)По локализации:
Генеративные или гаметические происходят в процессе образования половых клеток (нарушения мейоза)
2)По локализации:
Генеративные или гаметические происходят в процессе образования половых клеток (нарушения мейоза)


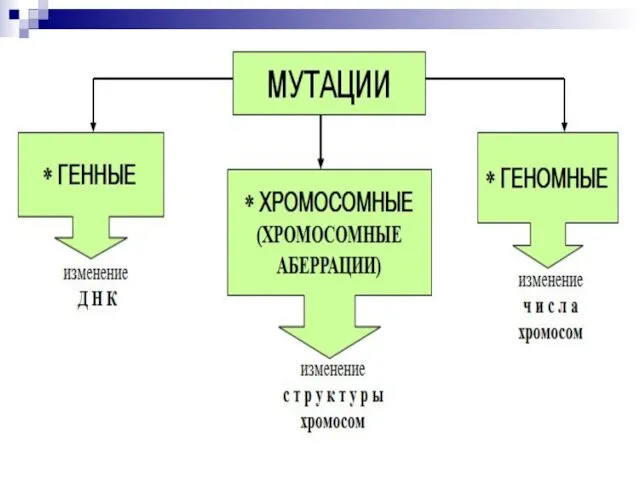
Слайд 44Все хромосомные болезни можно разделить на 2 группы:
Геномные, связанные с аномалиями числа
Все хромосомные болезни можно разделить на 2 группы:
Геномные, связанные с аномалиями числа

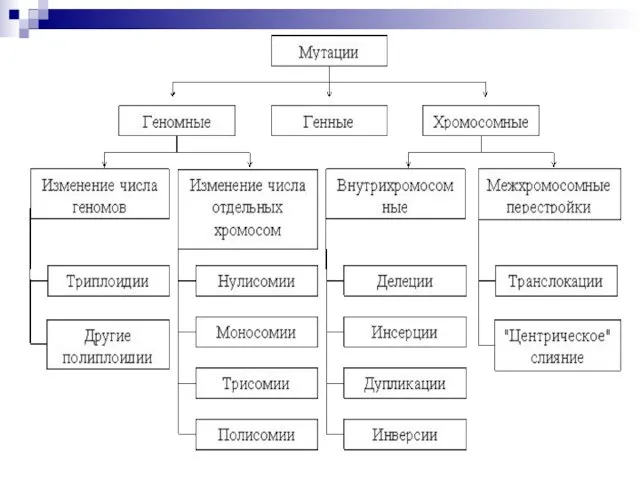
Слайд 464) По уровню организации наследственного аппарата:
Геномные мутации обусловлены изменением числа хромосом.
Причины:
а)нерасхождения хромосом,
4) По уровню организации наследственного аппарата:
Геномные мутации обусловлены изменением числа хромосом.
Причины:
а)нерасхождения хромосом,



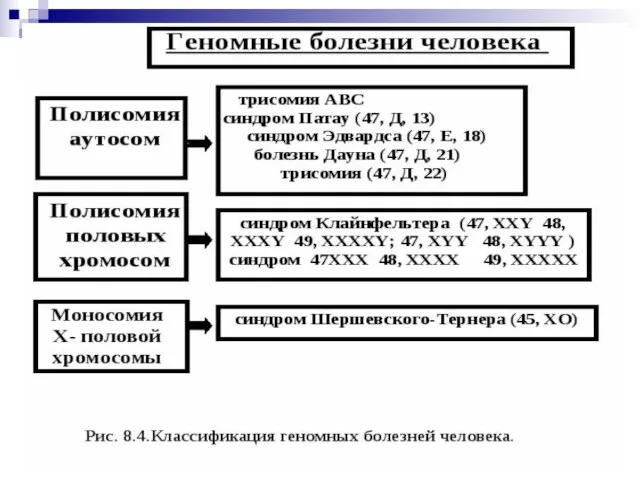
Слайд 49Гетероплоидия, или анеуплоидия - некратное гаплоидному уменьшение или увеличение числа хромосом (2n+1,
Гетероплоидия, или анеуплоидия - некратное гаплоидному уменьшение или увеличение числа хромосом (2n+1,

Слайд 50Гетероплоидия —изменение числа хромосом, не кратное гаплоидному набору. При этом набор хромосом
Гетероплоидия —изменение числа хромосом, не кратное гаплоидному набору. При этом набор хромосом

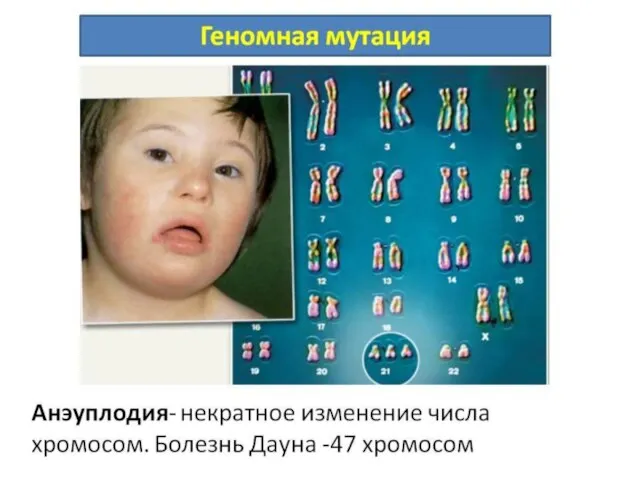
Слайд 52Новорожденный с синдромом Дауна отличается характерным внешним видом: округлый череп со скошенным
Новорожденный с синдромом Дауна отличается характерным внешним видом: округлый череп со скошенным

Слайд 53синдром Патау (трисомия по 13 паре аутосом), характеризующийся отсутствием шеи, различными уродствами
синдром Патау (трисомия по 13 паре аутосом), характеризующийся отсутствием шеи, различными уродствами

Слайд 58Аномалии половых хромосом
47, ХХХ – трисомия Х Частота 1:800, 1:1000. Фенотип
Аномалии половых хромосом
47, ХХХ – трисомия Х Частота 1:800, 1:1000. Фенотип


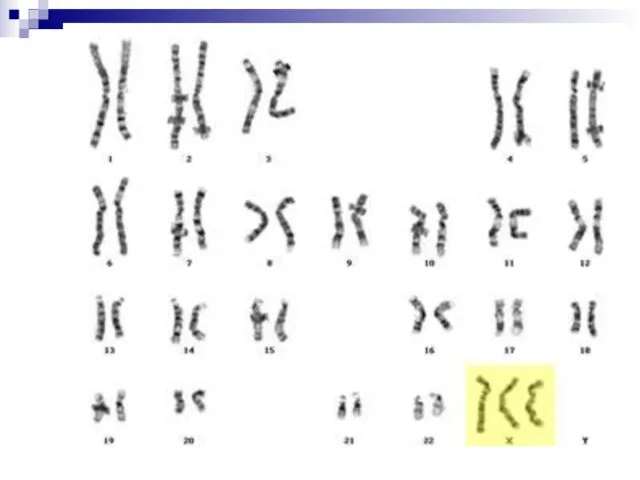
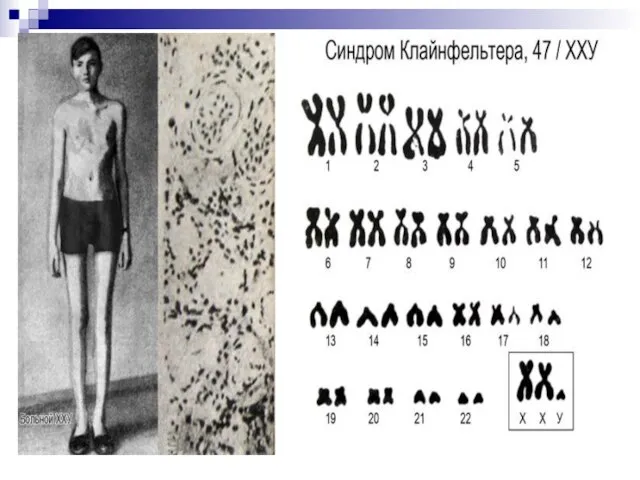
Слайд 6147, ХХY; 48, XXXY и др. – синдром Клайнфельтера. Частота – 1:400,
47, ХХY; 48, XXXY и др. – синдром Клайнфельтера. Частота – 1:400,

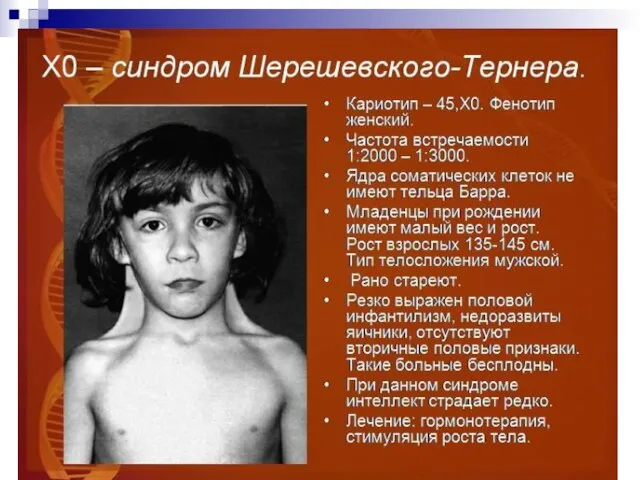
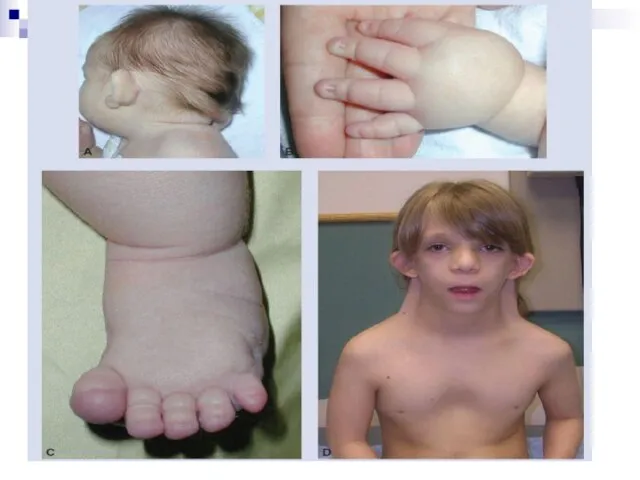
Слайд 65Помимо изменений в числе хромосом, известны и случаи изменений в структуре хромосом,
Помимо изменений в числе хромосом, известны и случаи изменений в структуре хромосом,


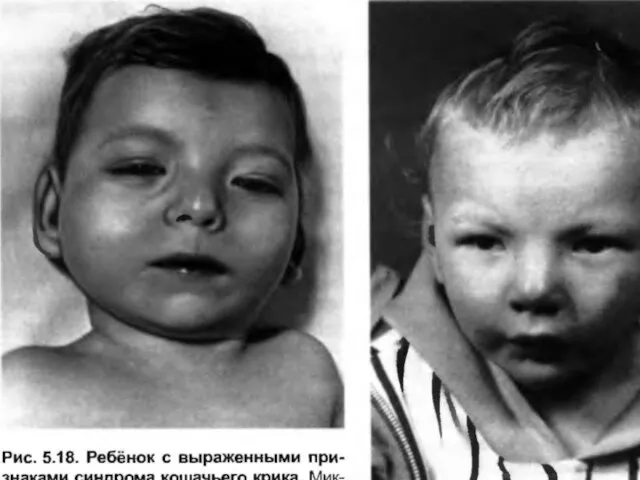
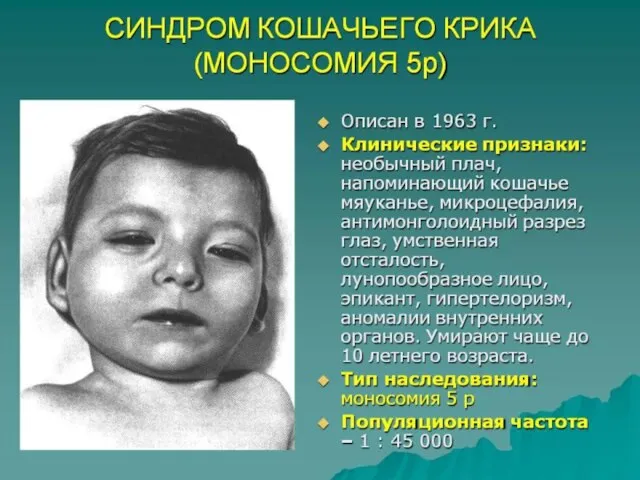

Слайд 70Хромосомная патология является одним из основных факторов формирования множественных пороков развития и
Хромосомная патология является одним из основных факторов формирования множественных пороков развития и

Слайд 71Хромосомные мутации (абберации) обусловлены изменением структуры хромосом.
К внутрихромосомным мутациям относятся перестройки внутри
Хромосомные мутации (абберации) обусловлены изменением структуры хромосом.
К внутрихромосомным мутациям относятся перестройки внутри

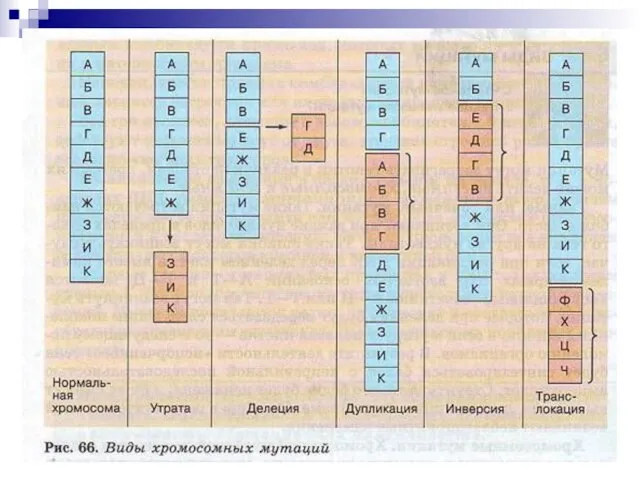
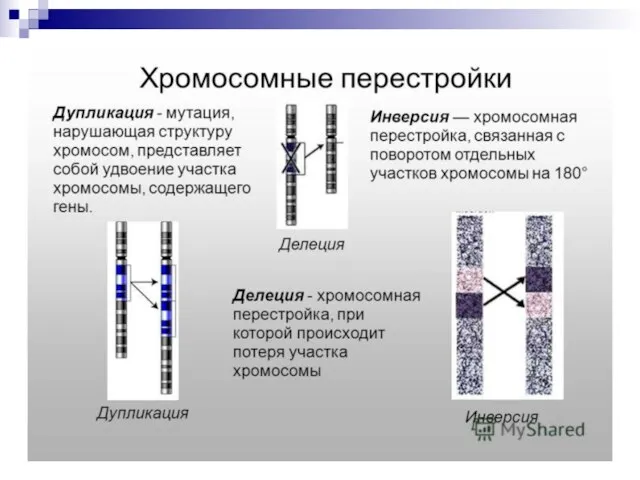
Слайд 74ХРОМОСОМНАЯ ТЕРМИНОЛОГИЯ
1. В самом начале указывается общее число хромосом: 46,45,47.
2.Состав половых хромосом: 46.ХХ -нормальный
ХРОМОСОМНАЯ ТЕРМИНОЛОГИЯ
1. В самом начале указывается общее число хромосом: 46,45,47.
2.Состав половых хромосом: 46.ХХ -нормальный

Слайд 75Генные (точковые) мутации связанны с изменением структуры гена (молекулы ДНК), могут затрагивать
Генные (точковые) мутации связанны с изменением структуры гена (молекулы ДНК), могут затрагивать

Слайд 76Генные мутации у человека являются причинами многих наследственных моногенных заболеваний. При изучении
Генные мутации у человека являются причинами многих наследственных моногенных заболеваний. При изучении

Слайд 77Фенотипически генные мутации проявляются как наследственные болезни обмена веществ - ферментопатии. Вещества,
Фенотипически генные мутации проявляются как наследственные болезни обмена веществ - ферментопатии. Вещества,

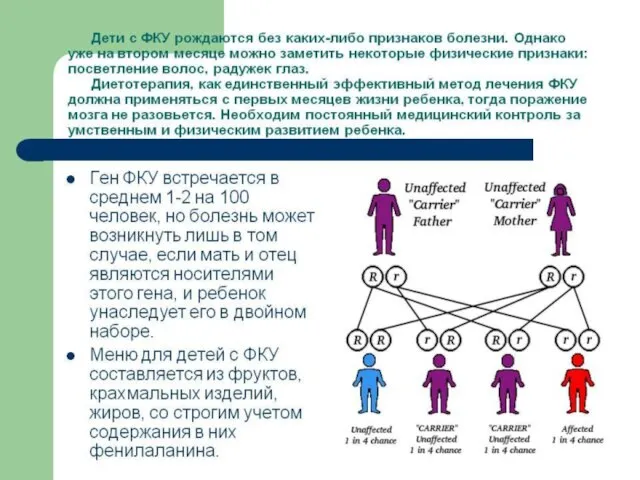
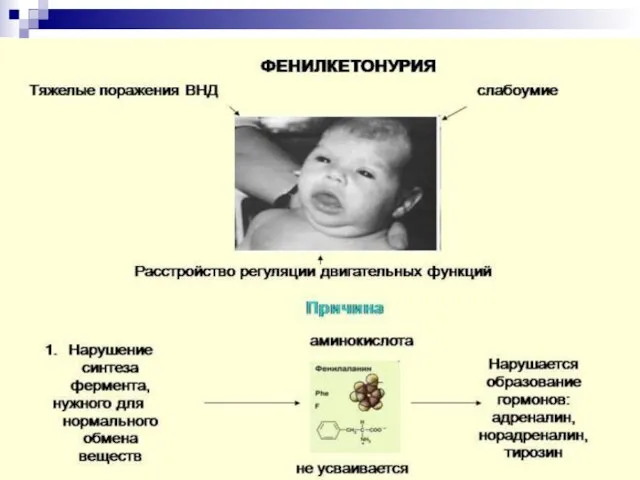
Слайд 78Наиболее часто встречающимися болезнями, связанными с нарушением аминокислотного обмена являются: фенилкетонурия и
Наиболее часто встречающимися болезнями, связанными с нарушением аминокислотного обмена являются: фенилкетонурия и

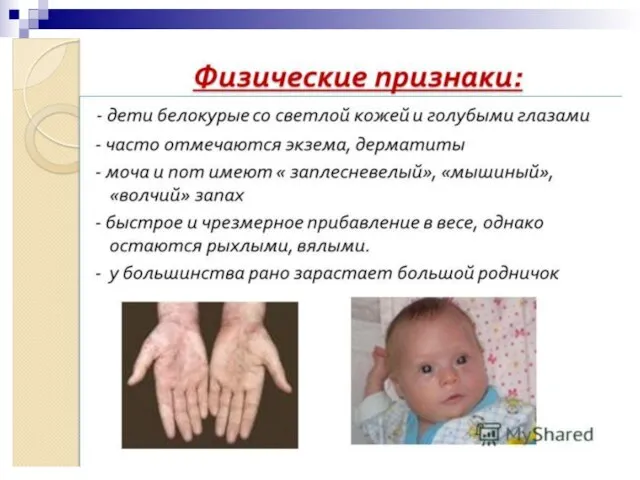
Слайд 80Альбинизм встречается в разных популяциях с разной частотой - от 1:5000 до
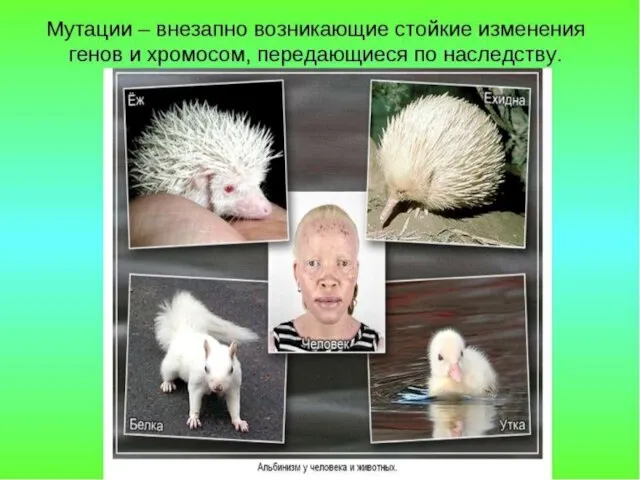
Альбинизм встречается в разных популяциях с разной частотой - от 1:5000 до

Слайд 82Алкаптонурия встречается довольно редко
(3-5:1000000). Наследуется по аутосомно-рецессивному
типу. Алкаптонурия является следствием генетического
дефекта
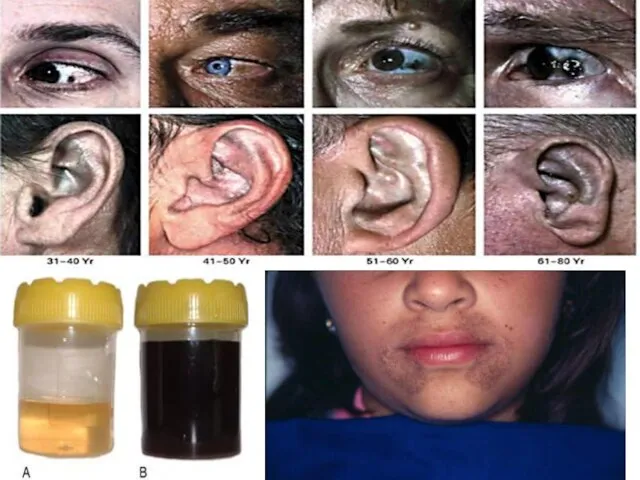
Алкаптонурия встречается довольно редко
(3-5:1000000). Наследуется по аутосомно-рецессивному
типу. Алкаптонурия является следствием генетического
дефекта

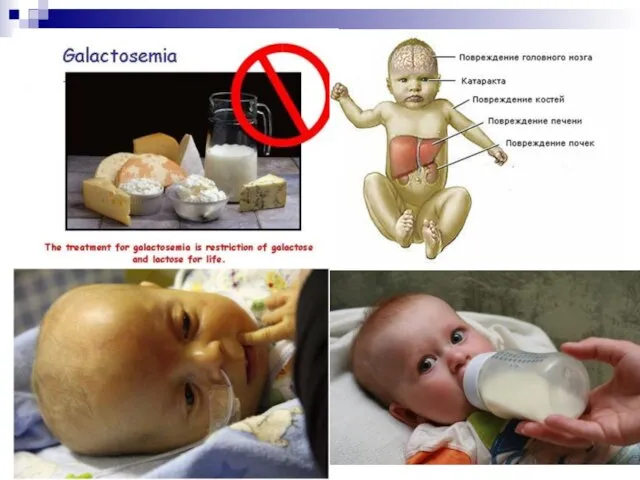
Слайд 84Наиболее частыми наследственными дефектами нарушения обмена углеводов являются галактоземия и мукополисахаридозы.
Галактоземия встречается
Наиболее частыми наследственными дефектами нарушения обмена углеводов являются галактоземия и мукополисахаридозы.
Галактоземия встречается

Слайд 86Наследственные дефекты обмена липидов : Эти
болезни встречаются редко (1:300000). Высокая частота
болезни Тея-Сакса
Наследственные дефекты обмена липидов : Эти
болезни встречаются редко (1:300000). Высокая частота
болезни Тея-Сакса

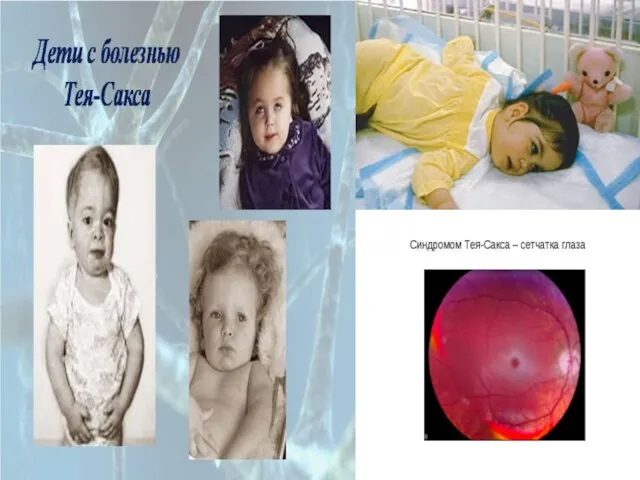
Слайд 88Гиперлипопротеинемии обусловлены нарушением обмена липидов плазмы крови вследствие дефектов ферментов или клеточных
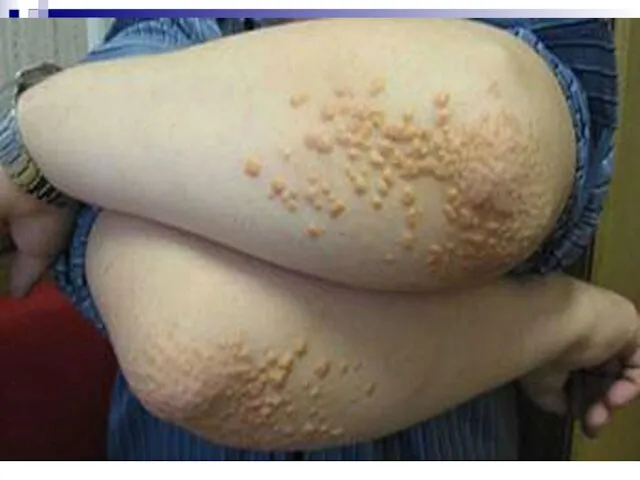
Гиперлипопротеинемии обусловлены нарушением обмена липидов плазмы крови вследствие дефектов ферментов или клеточных

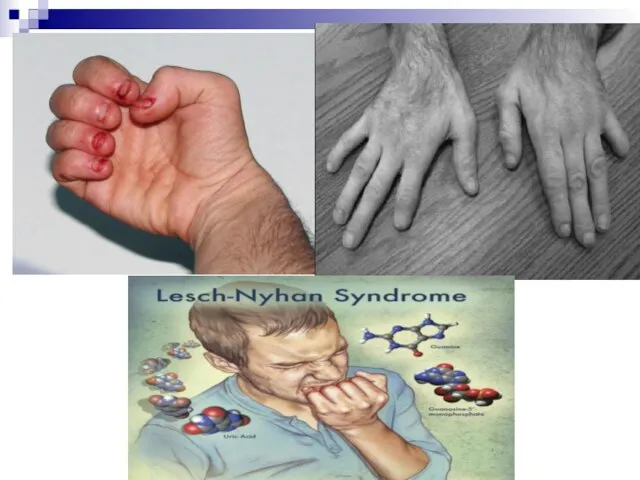
Слайд 90Наследственные дефекты обмена пуринов и пиримидинов
Синдром Леша-Нихана обусловлен недостаточностью фермента гипоксантин-фосфорибозилтрансферазы (ГФРТ), который катализирует присоединение свободных пуриновых
Наследственные дефекты обмена пуринов и пиримидинов
Синдром Леша-Нихана обусловлен недостаточностью фермента гипоксантин-фосфорибозилтрансферазы (ГФРТ), который катализирует присоединение свободных пуриновых

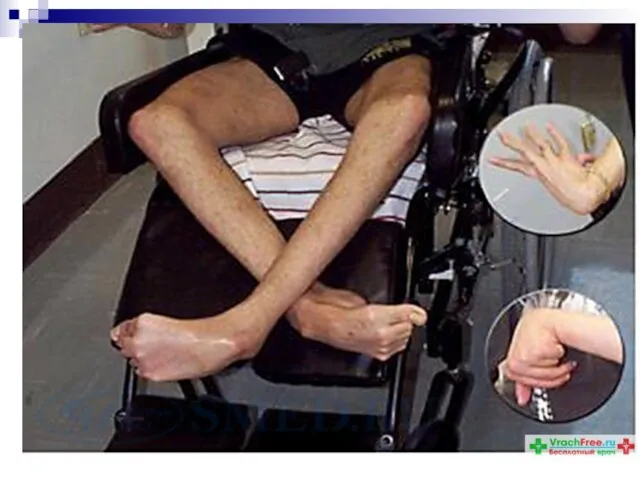
Слайд 92Примером нарушения минерального обмена может служить расстройство обмена меди.
Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия)
Примером нарушения минерального обмена может служить расстройство обмена меди.
Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия)


Слайд 95Наследственные заболевания , вызванные нарушением развития органов и тканей.
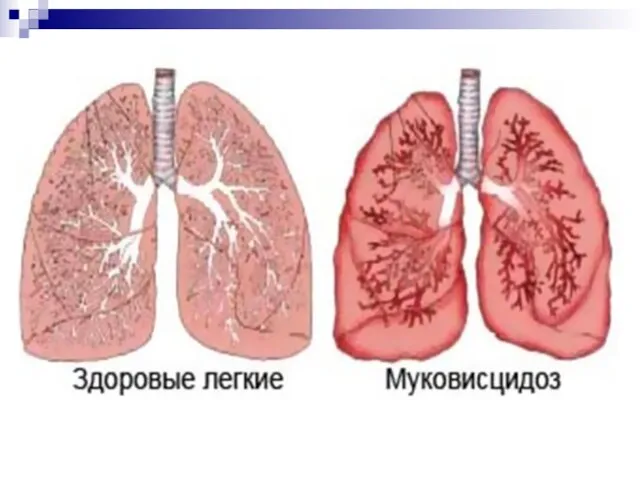
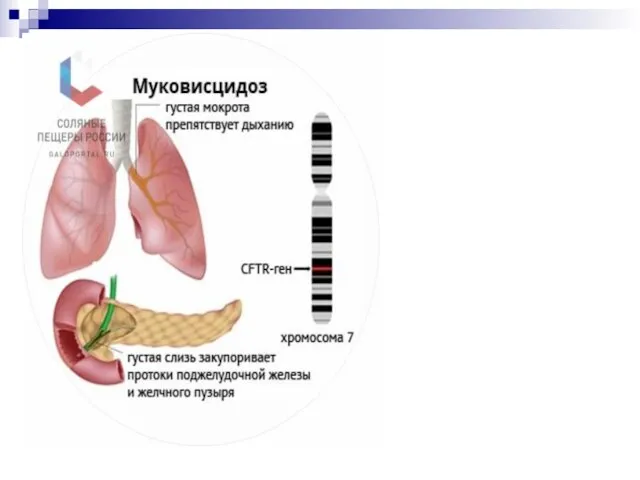
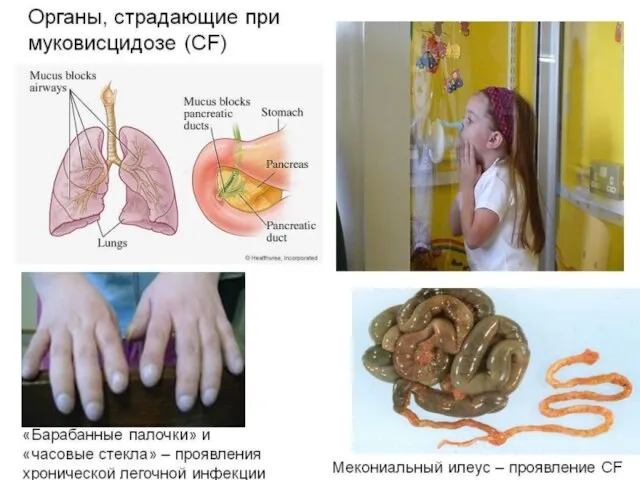
Муковисцидоз (кистофиброз поджелудочной железы)
Наследственные заболевания , вызванные нарушением развития органов и тканей.
Муковисцидоз (кистофиброз поджелудочной железы)

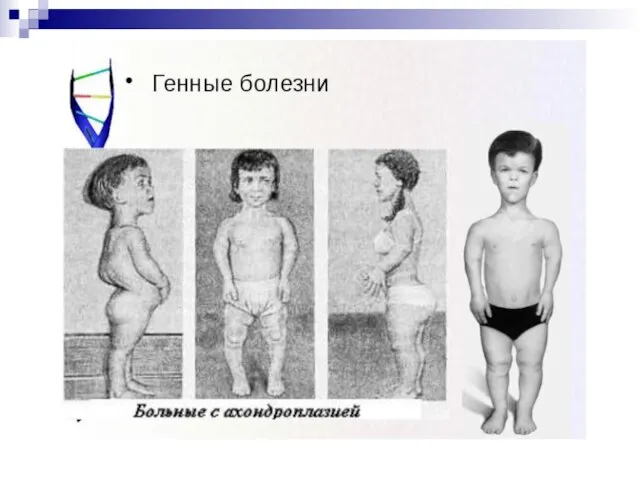
Слайд 99Ахондроплазия (хондродистрофия) обусловлена генной мутацией, вызывающей отклонения в активности некоторых ферментов (5-
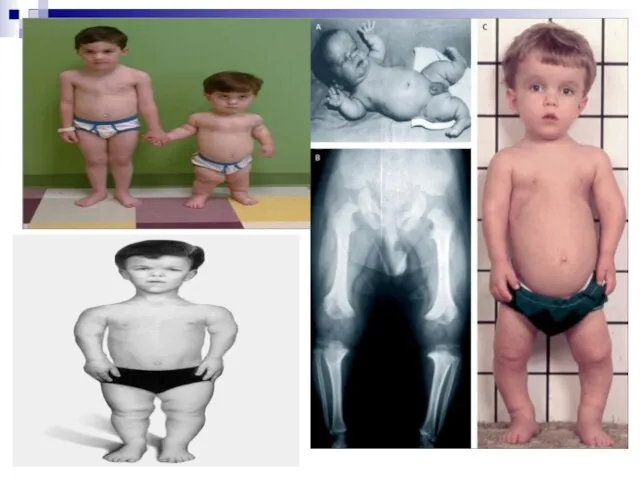
Ахондроплазия (хондродистрофия) обусловлена генной мутацией, вызывающей отклонения в активности некоторых ферментов (5-

Слайд 101Миодистрофия Дюшенна (МД) - тяжелое наследственное заболевание обусловленное мутацией гена дистрофина, что
Миодистрофия Дюшенна (МД) - тяжелое наследственное заболевание обусловленное мутацией гена дистрофина, что

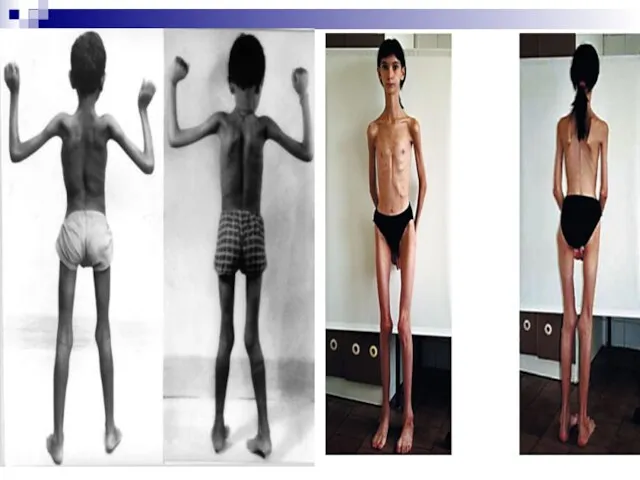
Слайд 103Синдром фрагильной (ломкой) Х-хромосомы - тяжелое наследственное заболевание. Тип наследования – Х-сцепленный
Синдром фрагильной (ломкой) Х-хромосомы - тяжелое наследственное заболевание. Тип наследования – Х-сцепленный

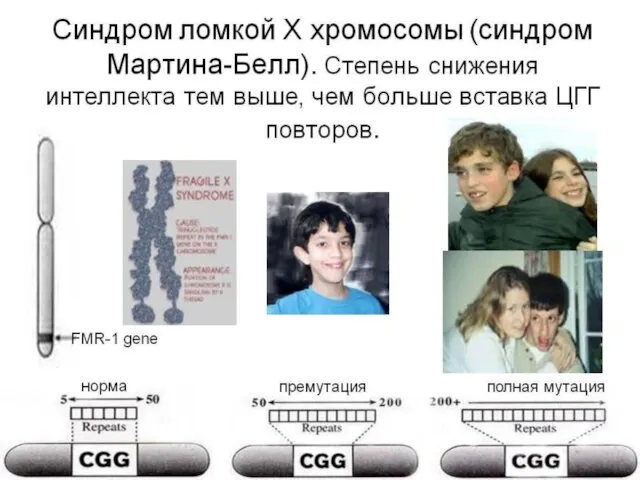
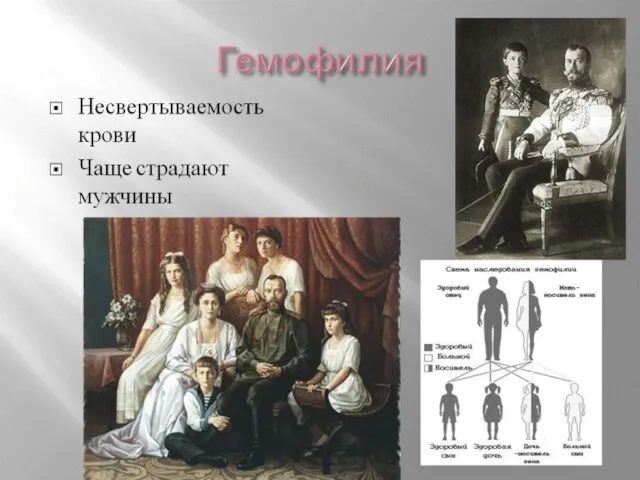
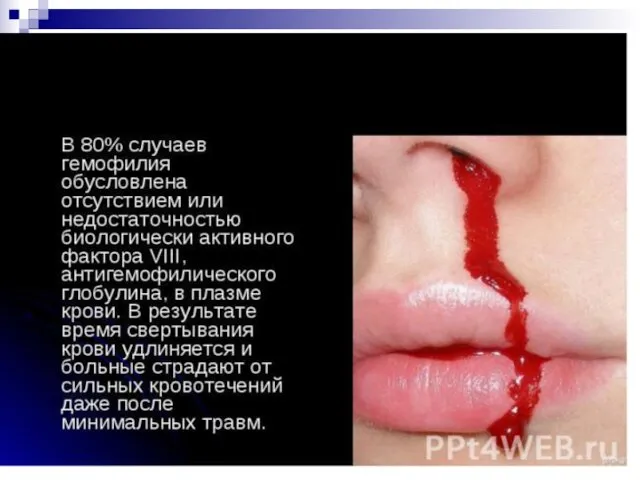
Слайд 105Наследственные заболевания нарушения свертывающей системы крови.
Гемофилия А - тяжелое наследственное заболевание, обусловленное
Наследственные заболевания нарушения свертывающей системы крови.
Гемофилия А - тяжелое наследственное заболевание, обусловленное

Слайд 106Гемоглобинопатии - заболевания, связанные с нарушением структуры молекулы гемоглобина. Нормальный гемоглобин человека
Гемоглобинопатии - заболевания, связанные с нарушением структуры молекулы гемоглобина. Нормальный гемоглобин человека

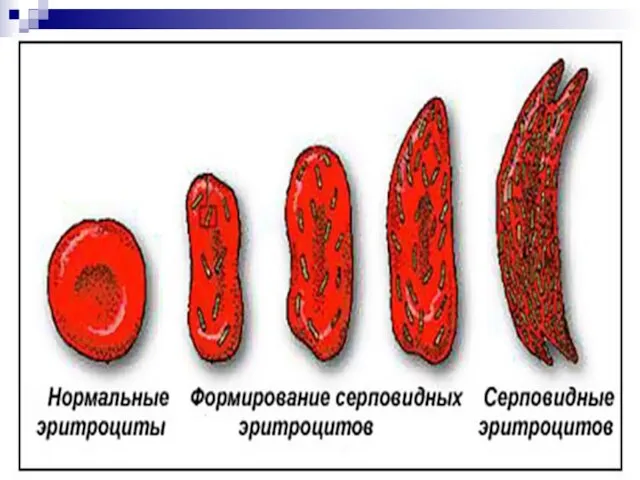
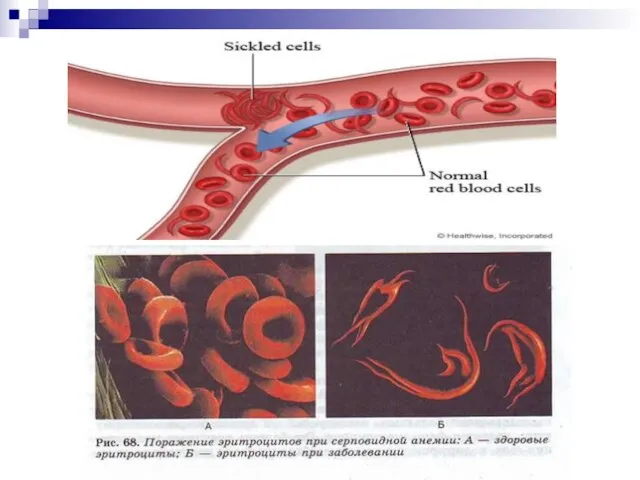
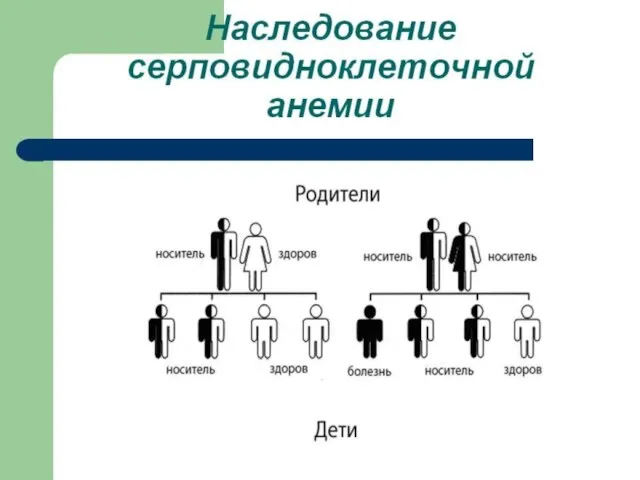
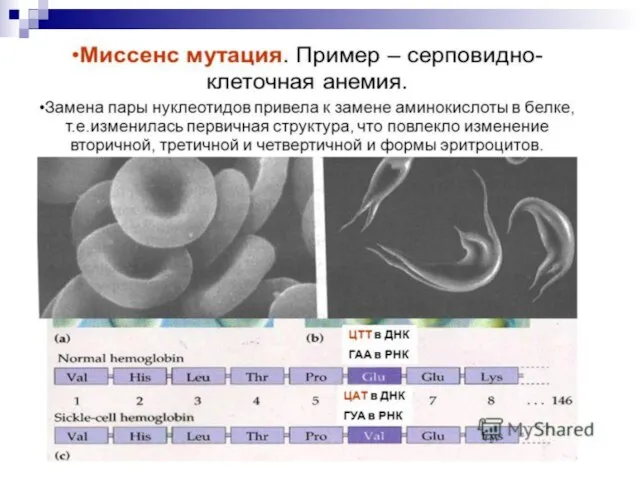





Слайд 121Повторим лекцию
Повторим лекцию

Слайд 122Виды наследования
Моногенное - тип наследования, при котором признак определяется ОДНИМ геном.
Полигенное -
Виды наследования
Моногенное - тип наследования, при котором признак определяется ОДНИМ геном.
Полигенное -

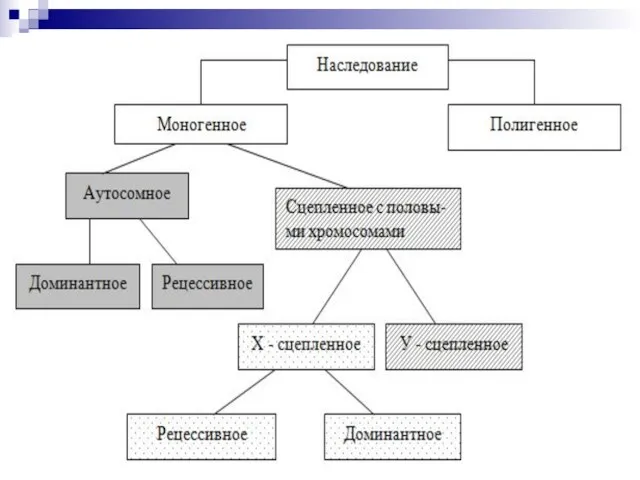
Слайд 124Моногенное наследование — наследование одного признака.
• аутомосный доминантный;
• аутосомный рецессивный;
• Х-сцепленный доминантный;
•
Моногенное наследование — наследование одного признака.
• аутомосный доминантный;
• аутосомный рецессивный;
• Х-сцепленный доминантный;
•

Слайд 125Доминантное наследование
Доминантное наследование — когда признак кодируется доминантным геном. Ген считается доминантным, если
Доминантное наследование
Доминантное наследование — когда признак кодируется доминантным геном. Ген считается доминантным, если

Слайд 126Примером доминантного наследования является наследование заболевания Хорея Гентингтона. Хорея Гентингтона — дегенеративное
Примером доминантного наследования является наследование заболевания Хорея Гентингтона. Хорея Гентингтона — дегенеративное

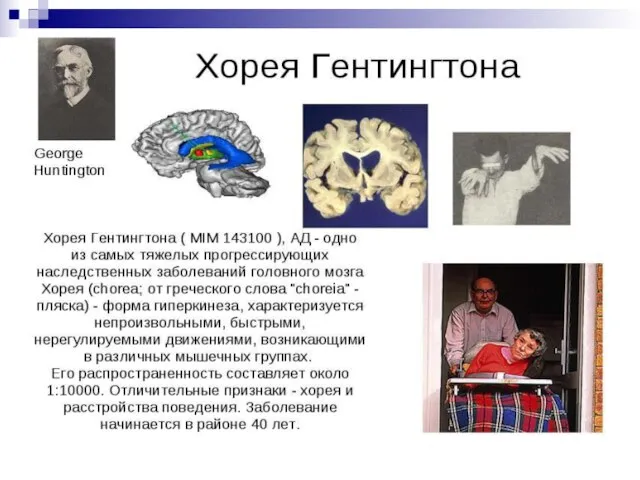
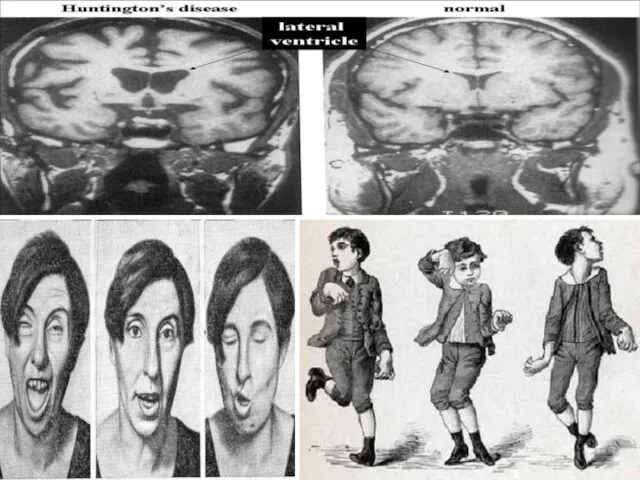
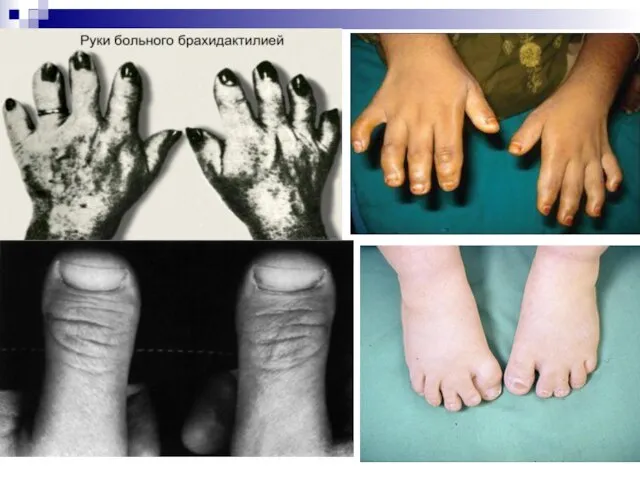
Слайд 129Ещё одним примером доминантного моногенного наследования может служить брахидактилия (короткопалость). Анализ семейных
Ещё одним примером доминантного моногенного наследования может служить брахидактилия (короткопалость). Анализ семейных

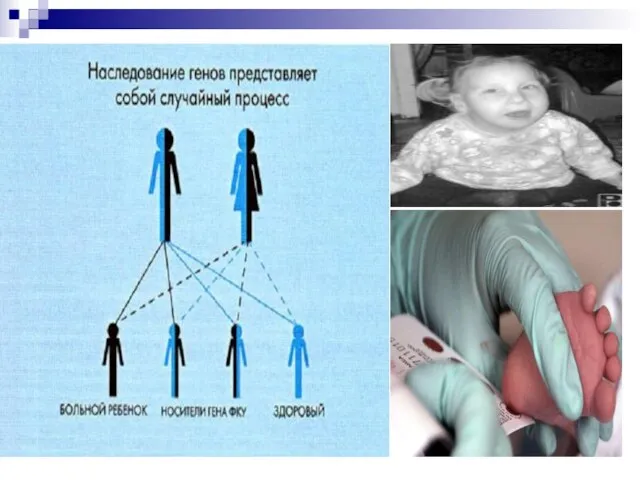
Слайд 131Рецессивное наследование.
Рецессивным ген считается, если признак, который он кодирует, не проявляется в
Рецессивное наследование.
Рецессивным ген считается, если признак, который он кодирует, не проявляется в

Слайд 132Особенности рецессивного наследования:
1. Признак проявляется только у рецессивных гомозиготных особей, при генотипе
Особенности рецессивного наследования: 1. Признак проявляется только у рецессивных гомозиготных особей, при генотипе



Слайд 139К менделеевскому наследованию по доминантному-рецессивному типу относится и наследование резус-фактора крови. Ген,
К менделеевскому наследованию по доминантному-рецессивному типу относится и наследование резус-фактора крови. Ген,

Слайд 140Резус-конфликт — это гуморальный иммунный ответ резус-отрицательной матери на эритроцитарные антигены резус-положительного
Резус-конфликт — это гуморальный иммунный ответ резус-отрицательной матери на эритроцитарные антигены резус-положительного

Слайд 141Варианты возникновения резус-конфликта:
• Если мать является резус-отрицательной, а отец — гомозиготным резус-положительным,
Варианты возникновения резус-конфликта:
• Если мать является резус-отрицательной, а отец — гомозиготным резус-положительным,

Слайд 142В подавляющем большинстве случаев резус-конфликт может быть предупреждён путём внутримышечного введения резус-отрицательной
В подавляющем большинстве случаев резус-конфликт может быть предупреждён путём внутримышечного введения резус-отрицательной

Слайд 143Неполное доминирование — в этом случае гетерозигота занимает промежуточное положение между доминантной и
Неполное доминирование — в этом случае гетерозигота занимает промежуточное положение между доминантной и

Слайд 144Кодоминирование — в фенотипе гетерозиготы проявляются два признака. Примером может являться наследование четвёртой
Кодоминирование — в фенотипе гетерозиготы проявляются два признака. Примером может являться наследование четвёртой

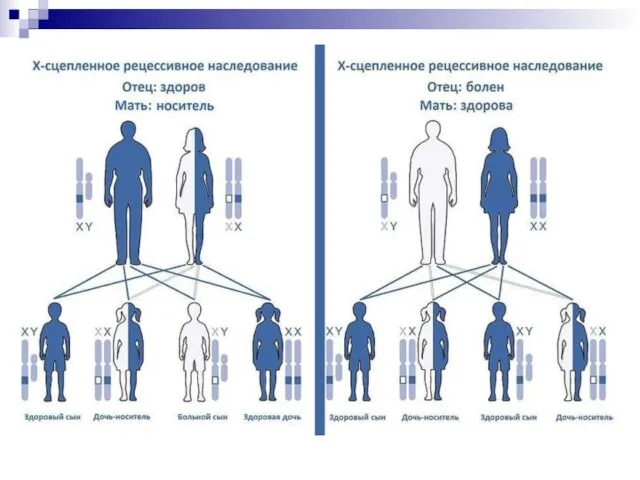
Слайд 145 Наследование, сцепленное с полом. Гены могут находиться на половых хромосомах, в этом случае
Наследование, сцепленное с полом. Гены могут находиться на половых хромосомах, в этом случае

Слайд 146Например, гемофилии — болезни, связанной с нарушением нормальной свёртываемости крови. При этих
Например, гемофилии — болезни, связанной с нарушением нормальной свёртываемости крови. При этих

Слайд 147Самой известной носительницей гемофилии в истории была королева Виктория; по-видимому, эта мутация
Самой известной носительницей гемофилии в истории была королева Виктория; по-видимому, эта мутация


Слайд 151Среди других генов, сцепленных с полом, стоит упомянуть гены, связанные с цветовой
Среди других генов, сцепленных с полом, стоит упомянуть гены, связанные с цветовой

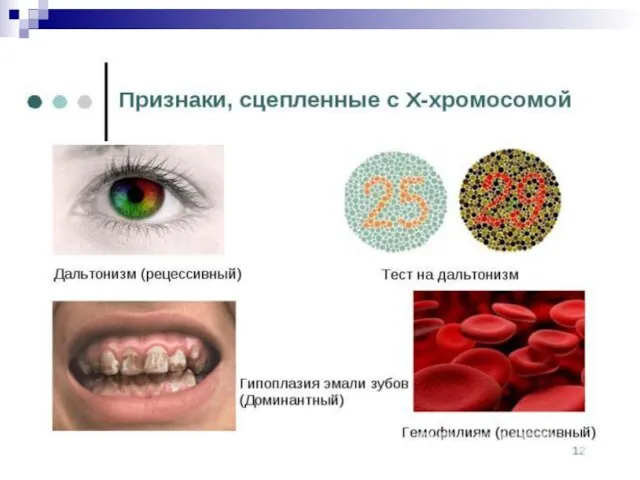
Слайд 153Встречаются и доминантные гены, сцепленные с Х-хромосомой Так, существует наследственная форма рахита,
Встречаются и доминантные гены, сцепленные с Х-хромосомой Так, существует наследственная форма рахита,

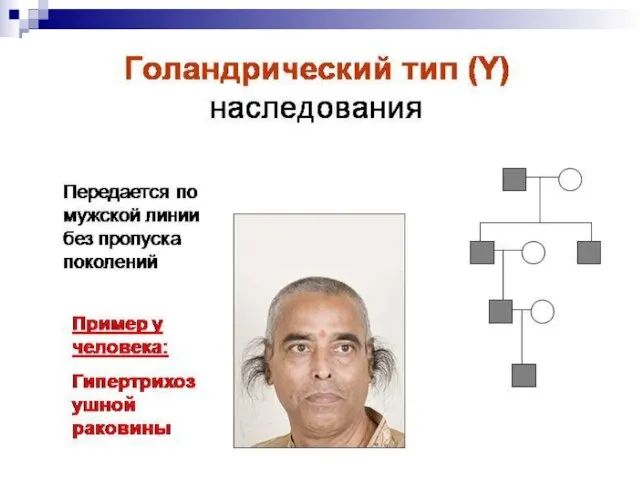
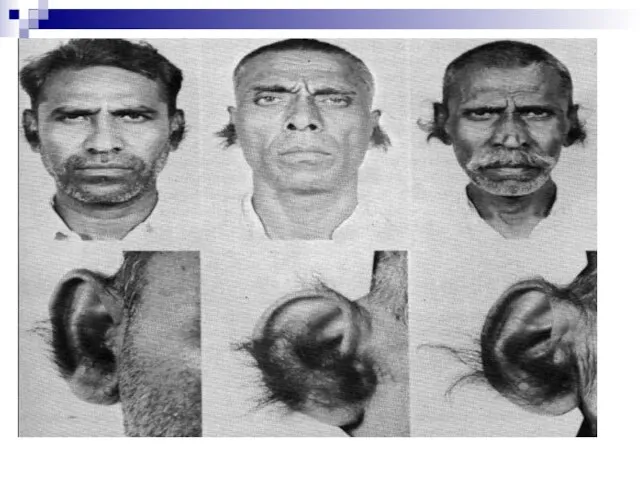
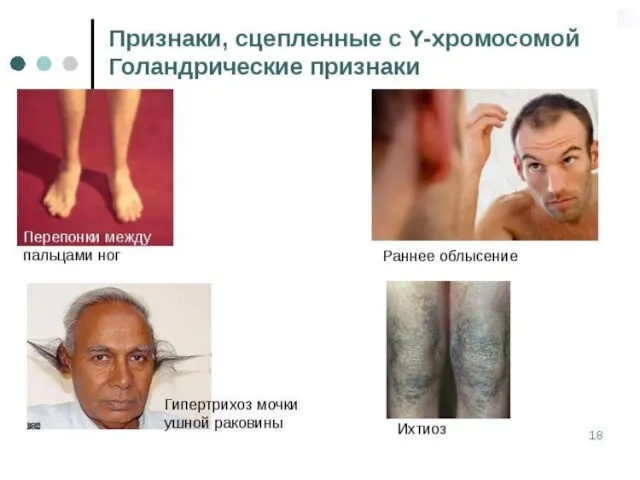
Слайд 157От наследования, сцепленного с полом, надо отличать наследование, ограниченное полом. В случае
От наследования, сцепленного с полом, надо отличать наследование, ограниченное полом. В случае


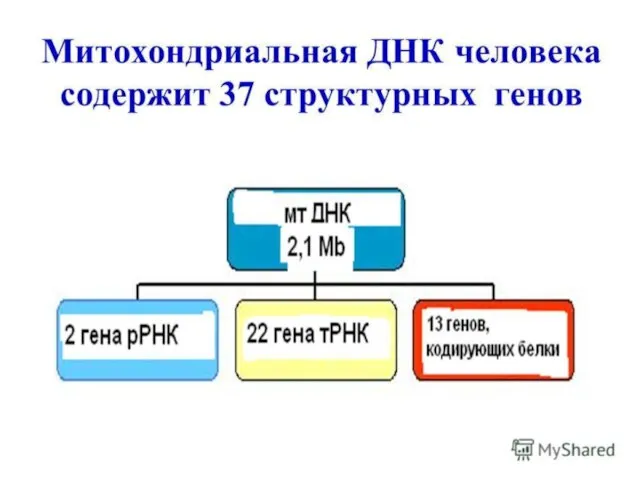
Слайд 159Митохондриальное наследование.
Митохондриальная ДНК представляет из себя одну кольцевидно-замкнутую хромосому. Закономерности митохондриального наследования:
•
Митохондриальное наследование.
Митохондриальная ДНК представляет из себя одну кольцевидно-замкнутую хромосому. Закономерности митохондриального наследования:
•


Слайд 162Полигенное наследование
Полигенное наследование — наследование признаков, зависящих от нескольких генов.
Полигенное наследование
Полигенное наследование — наследование признаков, зависящих от нескольких генов.

Слайд 163Комплементарность
такое взаимодействие генов, при котором 2 или более генов вызывают развитие признака.
Комплементарность
такое взаимодействие генов, при котором 2 или более генов вызывают развитие признака.

Слайд 164Полимерия
— несколько генов действуют на один признак одинаково. При этом при формировании
Полимерия
— несколько генов действуют на один признак одинаково. При этом при формировании

Слайд 165Плейотропия
Плейотропия — влияние одного гена на появление нескольких признаков. Примером может служить аутосомно-доминантное
Плейотропия
Плейотропия — влияние одного гена на появление нескольких признаков. Примером может служить аутосомно-доминантное

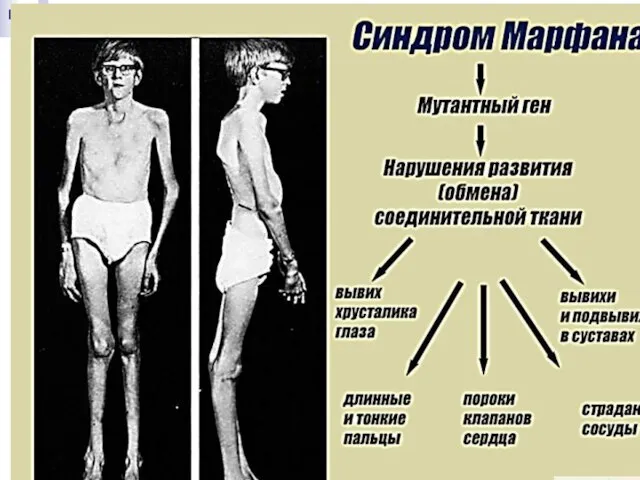
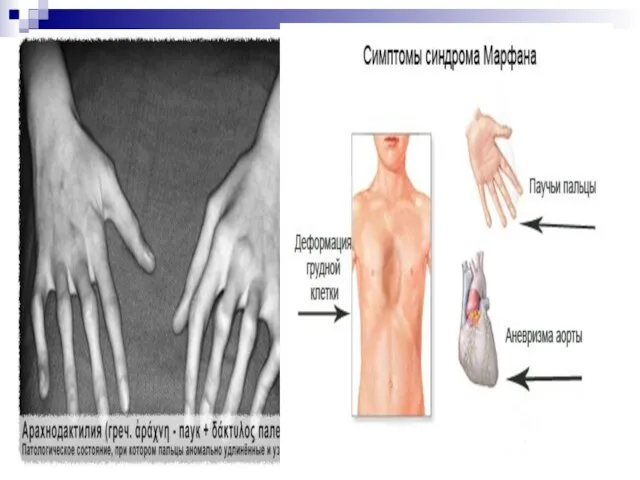
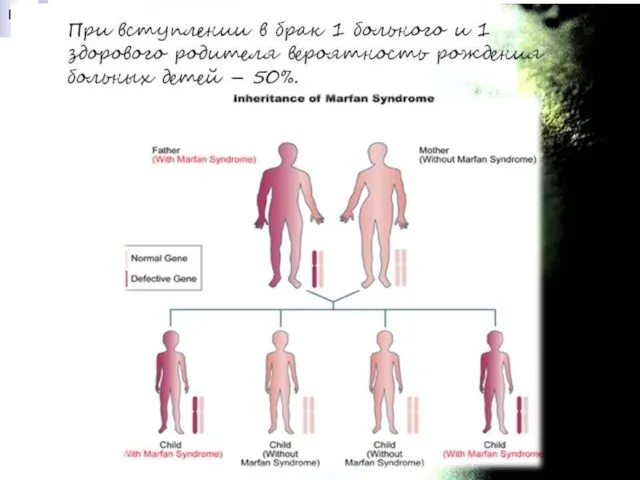
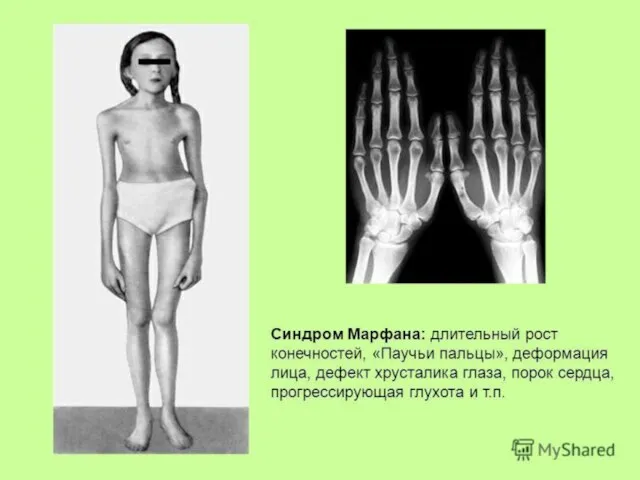
Слайд 170Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который

Слайд 171Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30-40 годами
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30-40 годами


Слайд 173Эпистаз
подавление одним геном другого, неаллельного. Примером эпистаза может служить "бомбейский феномен".
Эпистаз
подавление одним геном другого, неаллельного. Примером эпистаза может служить "бомбейский феномен".

Слайд 174Ещё одним примером эпистаза может служить появление белых альбиносов в семье темнокожих.
Ещё одним примером эпистаза может служить появление белых альбиносов в семье темнокожих.


Слайд 176Синдром Морриса
— синдром нечувствительности к андрогенам (синдром тестикулярной феминизации) проявляется нарушениями полового
Синдром Морриса
— синдром нечувствительности к андрогенам (синдром тестикулярной феминизации) проявляется нарушениями полового

Слайд 177Синдром тестикулярной феминизации (СТФ) впервые описан в 1817 году баварским врачом, он
Синдром тестикулярной феминизации (СТФ) впервые описан в 1817 году баварским врачом, он

Слайд 178СТФ очень редок — по данным разных авторов, частота его составляет от 1:65 000 до
СТФ очень редок — по данным разных авторов, частота его составляет от 1:65 000 до

Слайд 179 большинстве исследований пациентов с синдромом Морриса описана не только их физическая привлекательность,
большинстве исследований пациентов с синдромом Морриса описана не только их физическая привлекательность,

Слайд 180Великая «неженщины»:
7 особенностей, которые позволили утверждать, что у Жанны д'Арк был
Великая «неженщины»:
7 особенностей, которые позволили утверждать, что у Жанны д'Арк был

 Электронная торговля в России:география покупателей и доставки
Электронная торговля в России:география покупателей и доставки Brest. Brest Nord
Brest. Brest Nord Презентация на тему Техника безопасности на уроках физики
Презентация на тему Техника безопасности на уроках физики К вопросу о реабилитации больных после хирургического лечения ожирения. Кто, как и где должен обучать пациентов вопросам правильн
К вопросу о реабилитации больных после хирургического лечения ожирения. Кто, как и где должен обучать пациентов вопросам правильн Информационные технологии в промышленности и экономике
Информационные технологии в промышленности и экономике Форма государства. Гражданство
Форма государства. Гражданство Проанализируйте специфику функционирования и работы проектной команды
Проанализируйте специфику функционирования и работы проектной команды Образ И.И.Обломова в романе И.А.Гончарова «Обломов»
Образ И.И.Обломова в романе И.А.Гончарова «Обломов» Презентация на тему Holidays in Great Britain (Праздники в Великобритании)
Презентация на тему Holidays in Great Britain (Праздники в Великобритании) Презентация на тему Сказка о рыбаке и рыбке
Презентация на тему Сказка о рыбаке и рыбке Диагностические экспресс-тесты
Диагностические экспресс-тесты Семейные основы социальной толерантности.
Семейные основы социальной толерантности. Потолочные системы
Потолочные системы Зоны негабаритности
Зоны негабаритности Разряды местоимений
Разряды местоимений Элементы статистики и теории вероятностей в курсе математики основной школы
Элементы статистики и теории вероятностей в курсе математики основной школы Презентация на тему Птицы Красной книги Казахстана
Презентация на тему Птицы Красной книги Казахстана  Движение тела в гравитационном поле.
Движение тела в гравитационном поле. Критерии для статистики Почему они важны? Данные последнего сезона не были последовательными Игры с 2 атакующими передачами за вес
Критерии для статистики Почему они важны? Данные последнего сезона не были последовательными Игры с 2 атакующими передачами за вес ПРОФИЛЬ КОМПАНИИ 2010. 2 Страховая группа ERGO Основана в 1997 году слиянием четырех компаний – Victoria, DKV, D.A.S. и Hamburg-Mannheimer Представлена.
ПРОФИЛЬ КОМПАНИИ 2010. 2 Страховая группа ERGO Основана в 1997 году слиянием четырех компаний – Victoria, DKV, D.A.S. и Hamburg-Mannheimer Представлена. / Души частичку письма дарят нам…
/ Души частичку письма дарят нам… Как преодолеть фобии
Как преодолеть фобии Разъяснение для заявления о переводе средств КФ
Разъяснение для заявления о переводе средств КФ Приготовление воскресного обеда. 6 класс
Приготовление воскресного обеда. 6 класс История математики
История математики Добро пожаловать на встречу За чашкой чая. Гостеприимство
Добро пожаловать на встречу За чашкой чая. Гостеприимство Моя прабабушка Пелагея Алексеевна Трубицына - участник Великой Отечественной войны.
Моя прабабушка Пелагея Алексеевна Трубицына - участник Великой Отечественной войны. А.М. Герасимов После дождя
А.М. Герасимов После дождя