- Прионные болезни человека

Содержание
- 2. Изучение с 1933 г болезней овец завезенных из Германии на о.Исландия , 1954г – завершение В.
- 3. Продолжительный (месяцы и годы) инкубационный период Медленно прогрессирующий характер течения Необычность поражения органов и тканей Неизбежность
- 4. Свойства возбудителя ТГЭ, сходные с известными вирусами Способен проходить через бактериальные фильтры с диаметром пор от
- 5. Новый класс инфекционных частиц, лишенный генетического материала 1985г. С.Прузинер получил из мозга хомяка зараженного скрепи безнуклеиновый
- 6. Поскольку было показано, что инфекционный прионный белок отличается от обычного третичной структурой, т.е. конформационно, вскоре появилось
- 7. Развитие медицинской вирусологии и молекулярной биологии позволило сделать следующий шаг в изучении медленных инфекций в период
- 8. Таким образом, был идентифицирован новый класс инфекционных агентов, и значение этого открытия сопоставимо по своей важности
- 9. Медленные инфекции ЦНС — группа инфекционных болезней человека и животных, вызываемых вирусами и прионами, характеризующихся медленным
- 10. Причины возникновения прионных болезней 1.Соматическая мутация PRNP или спонтанная конверсия PrPc в PrPSc при спорадических прионных
- 11. Процесс накопления молекул инфекционного прионного белка (“затравочная модель”) PrPsc PrP Трансгенные мыши, у которых отсутствовал ген
- 12. Скорость репродукции прионов Конверсия нормального белка PrPc cвязана с его синтезом. В течение 1ч в клетке
- 14. Предполагаемое изменение характера укладки полипептидной цепи при превращении белка РгРC (а) в прион РгРSc (б)
- 15. Прионные болезни человека В настоящее время к ним относят 4 болезни человека: болезнь Крейцфельда—Якоба, Куру, синдром
- 17. Ганс Герхард Крейтцфельдт (1885—1964) Спорадическая болезнь Крейтцфельдата-Якоба (сБКЯ) Спорадическими называют отдельные, разрозненные, эпидемиологически не связанные ни
- 18. Продромальные симптомы отмечаются у 1/3 больных за несколько недель или месяцев до появления кардинального признака БКЯ
- 19. Классическим клиническим проявлением БКЯ является прогрессирующая деменция (интеллектуальные и поведенческие нарушения, которые быстро нарастают) На ЭЭГ
- 20. Наследственные прионные болезни Обнаружено более 15 наиболее изученных мутаций в гене, кодирующем клеточный аналог прионов, в
- 21. Головной мозг человека, погибшего от болезни Крейтцфельдта-Якоба. Видны явные патоморфологические изменения: уменьшение объема и массы мозга,
- 22. 1- микровакуоли, 2 - погибающие нейроны с глиальными узелками, 3 - гипертрофия астроцитов, 4 - сморщенный
- 23. некоторые вакуоли слились и образовали более крупные полости, нейроны уже погибли. Увел. ´ 100. Микрофотография конечной
- 24. Семейная смертельная бессоница (ССМ), (синон. фатальная семейная инсомния - ФСИ) Впервые описана в 1986г. ССМ редкое
- 25. Синдром Герстмана-Штреусслера-Шейнкера ГШШ – редкое семейное заболевание, относящееся к генетически обусловленным формам спонгиозных энцефалопатий с аутосомно-доминантным
- 26. Приобретенные прионные болезни Куру Новый вариант БКЯ Ятрогенная БКЯ
- 27. Достоверно доказанное приобретенное воздействие прионов на человека Эпидемия Куру, связанная с ритуальным каннибализмом Эпизоотия спонгиоформной энцефалопатии
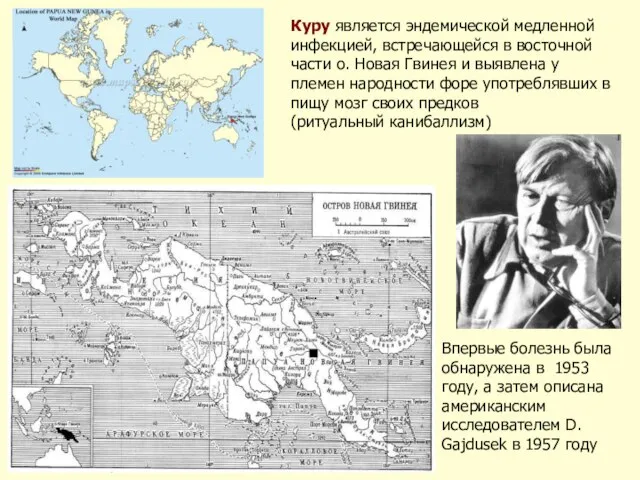
- 28. Куру является эндемической медленной инфекцией, встречающейся в восточной части о. Новая Гвинея и выявлена у племен
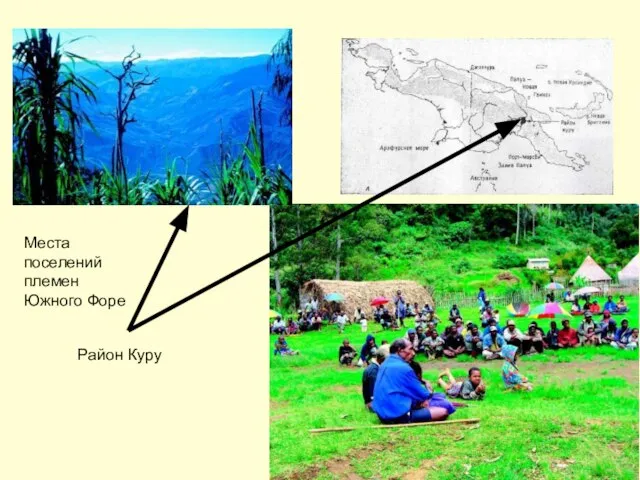
- 29. Район Куру Места поселений племен Южного Форе
- 30. В 1950-60 годах эпидемия Куру убила около 25% женской части популяции племен Южного Форе; в некоторых
- 31. Характеристики больных КУРУ, обследованных, в течение последних 15 лет Дата рождения Начало болезни возраст Смерть, возраст
- 32. КУРУ Куру, была первой болезнью из группы прионных инфекционность которой была доказана экспериментальным заражением обезьян биологическим
- 33. Новый вариант болезни Крейтцфельдата-Якоба (нв БКЯ) С 1994г. в Великобритании и затем во Франции стали регистрироваться
- 34. Инкубационный период развития нв БКЯ – от 1 года до 20 лет Как при новом, так
- 35. Случаи (указаны числами) «коровьего бешенства» среди сельскохозяйственных животных и риск заражения человека болезнью Крейтцфельдта-Якоба в некоторых
- 36. По степени вероятности заражения наиболее опасно употребление в пищу в первую очередь мозга и глаз, затем
- 37. При попадании в желудочно-кишечный тракт, прионы инфицируют дендритные клетки пейеровых бляшек, затем дендритные клетки “заселяют” лимфатические
- 38. В настоящее время полностью установлена связь между нв БКЯ и ГЭК, так как по трем генетическим
- 39. По данным CDC (США) с момента регистрации первого случая нового варианта болезни Крейтцфельдата-Якоба до августа 2006г
- 40. Ятрогенная БКЯ Ятрогенная БКЯ может рассматривается как внутрибольничная инфекция. Все выявленные случаи ятрогенных прионных болезней классифицируются
- 41. Гормон человеческого роста из гипофиза трупов применялся для лечения детей от карликовости с конца 1950-х до
- 42. Первый случай в мире был зарегистрирован как “возможная БКЯ” у реципиента в Великобритании, в декабре 2003г.
- 43. Случай 1. Пациент – мужчина 62-х лет получил переливание крови в 1997г при оперативном вмешательстве и
- 44. В июле 2004г английское правительство подтвердило наличие второго в Великобритании случая БКЯ после переливания крови. Второй
- 45. Последний (третий) случай заболевания зарегистрирован в декабре 2006г у молодого человека, 32-х лет получившего массивную трансфузию
- 46. По результатам опубликованного в 2006г эпидемиологического исследования (Hewitt PE, 2006г) 24 пациента с различными формами БКЯ
- 47. При заражении путем переливания крови регистрируется более короткий инкубационный период болезни, чем при заражении алиментрным путем
- 48. Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M,
- 49. В 2004г в Великобритании было принято решение исключить из донорства всех лиц, кто получил переливание крови
- 50. Донорской кровью, заготовленной в Великобритании, пользовались 11 стран мира, и исключить уже реализовавшийся риск заражения иностранных
- 51. Великобритания импортировала в 2005г кровь из США на сумму 116 миллионов долларов в связи с невозможностью
- 52. В связи с необходимостью прижизненной диагностики прионных болезней у лиц, находящихся в инкубационном периоде необходима экстренная
- 53. Дифференциальный диагноз болезни Крейтцфельдта – Якоба проводится со всеми болезнями, одним из проявлений которых является деменция:
- 54. Биологические методы диагностики Трансгенные мыши , несущие ген, кодирующий нормальный PrP человека рекомендованы ВОЗ для тестирования
- 55. Профилактика: Исключение способов проникновения возбудителя в организм (ограничено применение препаратов, изготовленных из тканей крупного рогатого скота,
- 56. ВОЗ, на современном этапе рекомендует 3 типа обработки не одноразового медицинского инструментария: физическая обработка: автоклавирование при
- 58. Скачать презентацию

Слайд 3 Продолжительный (месяцы и годы) инкубационный период
Медленно прогрессирующий характер течения
Необычность
Продолжительный (месяцы и годы) инкубационный период
Медленно прогрессирующий характер течения
Необычность

Слайд 4Свойства возбудителя ТГЭ, сходные с известными вирусами
Способен проходить через бактериальные фильтры
Свойства возбудителя ТГЭ, сходные с известными вирусами
Способен проходить через бактериальные фильтры

Слайд 5Новый класс инфекционных частиц,
лишенный генетического материала
1985г. С.Прузинер получил из мозга хомяка
Новый класс инфекционных частиц,
лишенный генетического материала
1985г. С.Прузинер получил из мозга хомяка

Слайд 6Поскольку было показано, что инфекционный прионный белок отличается от обычного третичной структурой,
Поскольку было показано, что инфекционный прионный белок отличается от обычного третичной структурой,

Слайд 7Развитие медицинской вирусологии и молекулярной биологии позволило сделать следующий шаг в изучении
Развитие медицинской вирусологии и молекулярной биологии позволило сделать следующий шаг в изучении

Слайд 8Таким образом, был идентифицирован новый класс инфекционных агентов,
и значение этого открытия
Таким образом, был идентифицирован новый класс инфекционных агентов,
и значение этого открытия

Слайд 9Медленные инфекции ЦНС — группа инфекционных болезней человека и животных, вызываемых вирусами
Медленные инфекции ЦНС — группа инфекционных болезней человека и животных, вызываемых вирусами

Слайд 10Причины возникновения прионных болезней
1.Соматическая мутация PRNP или спонтанная конверсия PrPc в PrPSc
Причины возникновения прионных болезней
1.Соматическая мутация PRNP или спонтанная конверсия PrPc в PrPSc

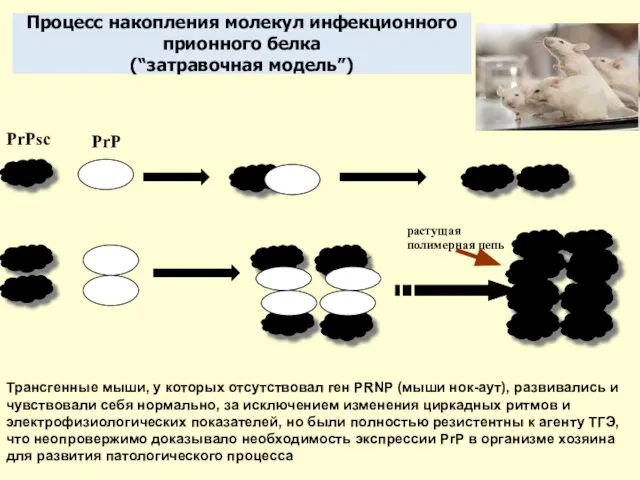
Слайд 11Процесс накопления молекул инфекционного
прионного белка
(“затравочная модель”)
PrPsc
PrP
Трансгенные мыши, у которых отсутствовал
Процесс накопления молекул инфекционного
прионного белка
(“затравочная модель”)
PrPsc
PrP
Трансгенные мыши, у которых отсутствовал
Слайд 12Скорость репродукции прионов
Конверсия нормального белка PrPc cвязана с его синтезом. В
Скорость репродукции прионов
Конверсия нормального белка PrPc cвязана с его синтезом. В

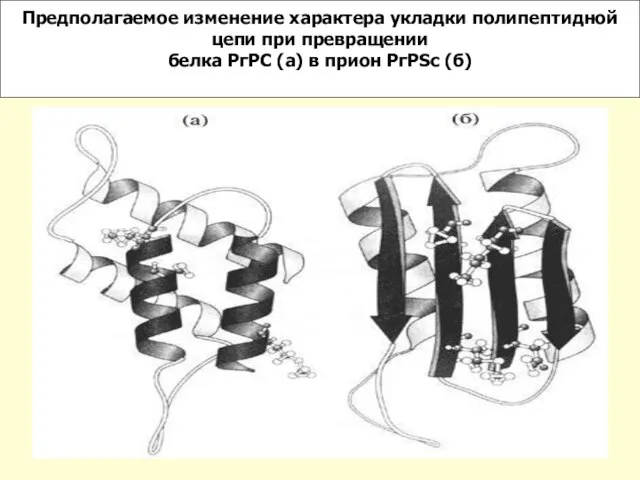
Слайд 14Предполагаемое изменение характера укладки полипептидной цепи при превращении
белка РгРC (а) в
Предполагаемое изменение характера укладки полипептидной цепи при превращении
белка РгРC (а) в
Слайд 15Прионные болезни человека
В настоящее время к ним относят 4 болезни человека:
болезнь Крейцфельда—Якоба,
Куру,
синдром
Прионные болезни человека
В настоящее время к ним относят 4 болезни человека:
болезнь Крейцфельда—Якоба,
Куру,
синдром


Слайд 17Ганс Герхард
Крейтцфельдт
(1885—1964)
Спорадическая болезнь Крейтцфельдата-Якоба (сБКЯ)
Спорадическими называют отдельные, разрозненные, эпидемиологически
Ганс Герхард
Крейтцфельдт
(1885—1964)
Спорадическая болезнь Крейтцфельдата-Якоба (сБКЯ)
Спорадическими называют отдельные, разрозненные, эпидемиологически

Слайд 18Продромальные симптомы отмечаются у 1/3 больных
за несколько недель или месяцев до
за несколько недель или месяцев до

Слайд 19Классическим клиническим проявлением БКЯ является прогрессирующая деменция
(интеллектуальные и поведенческие нарушения,
которые
Классическим клиническим проявлением БКЯ является прогрессирующая деменция
(интеллектуальные и поведенческие нарушения,
которые

Слайд 20Наследственные прионные болезни
Обнаружено более 15 наиболее изученных мутаций в гене, кодирующем
Наследственные прионные болезни
Обнаружено более 15 наиболее изученных мутаций в гене, кодирующем

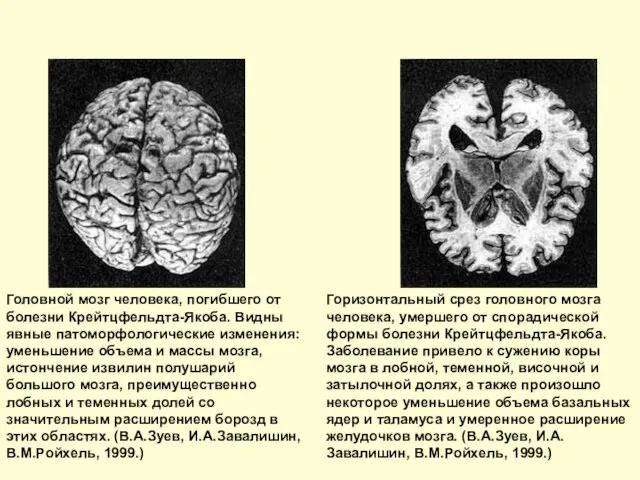
Слайд 21Головной мозг человека, погибшего от болезни Крейтцфельдта-Якоба. Видны явные патоморфологические изменения: уменьшение
Головной мозг человека, погибшего от болезни Крейтцфельдта-Якоба. Видны явные патоморфологические изменения: уменьшение
Слайд 221- микровакуоли, 2 - погибающие нейроны с глиальными узелками,
3 - гипертрофия
1- микровакуоли, 2 - погибающие нейроны с глиальными узелками,
3 - гипертрофия
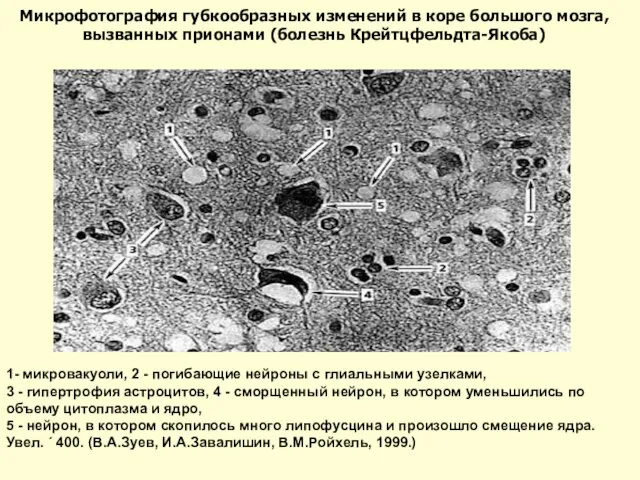
Слайд 23некоторые вакуоли слились и образовали более крупные полости, нейроны уже погибли. Увел.
некоторые вакуоли слились и образовали более крупные полости, нейроны уже погибли. Увел.

Слайд 24Семейная смертельная бессоница (ССМ),
(синон. фатальная семейная инсомния - ФСИ)
Впервые описана
Семейная смертельная бессоница (ССМ),
(синон. фатальная семейная инсомния - ФСИ)
Впервые описана

Слайд 25Синдром Герстмана-Штреусслера-Шейнкера
ГШШ – редкое семейное заболевание, относящееся к генетически обусловленным формам
Синдром Герстмана-Штреусслера-Шейнкера
ГШШ – редкое семейное заболевание, относящееся к генетически обусловленным формам

Слайд 26Приобретенные прионные болезни
Куру
Новый вариант БКЯ
Ятрогенная БКЯ
Приобретенные прионные болезни
Куру
Новый вариант БКЯ
Ятрогенная БКЯ
Слайд 27Достоверно доказанное приобретенное воздействие прионов на человека
Эпидемия Куру, связанная с ритуальным каннибализмом
Эпизоотия
Достоверно доказанное приобретенное воздействие прионов на человека
Эпидемия Куру, связанная с ритуальным каннибализмом
Эпизоотия

Слайд 28Куру является эндемической медленной инфекцией, встречающейся в восточной части о. Новая Гвинея
Куру является эндемической медленной инфекцией, встречающейся в восточной части о. Новая Гвинея
Слайд 29Район Куру
Места поселений племен
Южного Форе
Район Куру
Места поселений племен
Южного Форе
Слайд 30В 1950-60 годах эпидемия Куру убила около 25% женской части популяции племен
В 1950-60 годах эпидемия Куру убила около 25% женской части популяции племен

Слайд 31Характеристики больных КУРУ, обследованных, в течение последних 15 лет
Дата
рождения
Начало болезни
возраст
Смерть, возраст
Длит.
Характеристики больных КУРУ, обследованных, в течение последних 15 лет
Дата
рождения
Начало болезни
возраст
Смерть, возраст
Длит.
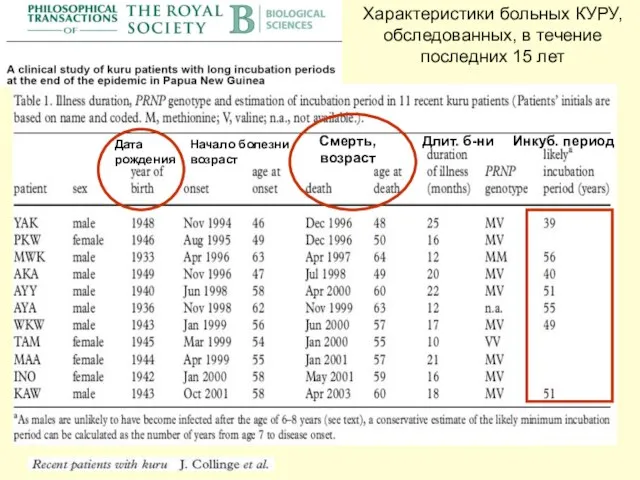
Слайд 32КУРУ
Куру, была первой болезнью из группы прионных инфекционность которой была доказана
КУРУ
Куру, была первой болезнью из группы прионных инфекционность которой была доказана

Слайд 33 Новый вариант болезни Крейтцфельдата-Якоба (нв БКЯ)
С 1994г. в Великобритании и
Новый вариант болезни Крейтцфельдата-Якоба (нв БКЯ)
С 1994г. в Великобритании и

Слайд 34 Инкубационный период развития нв БКЯ – от 1 года до 20
Инкубационный период развития нв БКЯ – от 1 года до 20

Слайд 35Случаи (указаны числами) «коровьего бешенства» среди сельскохозяйственных животных и риск заражения человека
Случаи (указаны числами) «коровьего бешенства» среди сельскохозяйственных животных и риск заражения человека
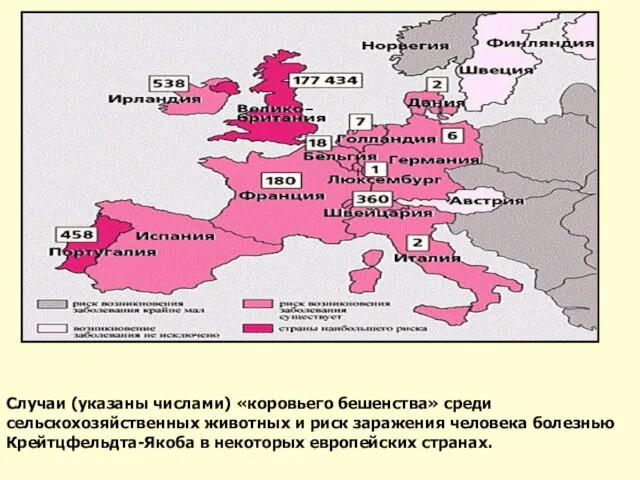
Слайд 36По степени вероятности заражения наиболее опасно употребление в пищу в первую очередь
По степени вероятности заражения наиболее опасно употребление в пищу в первую очередь

Слайд 37При попадании в желудочно-кишечный тракт, прионы инфицируют дендритные клетки пейеровых бляшек, затем
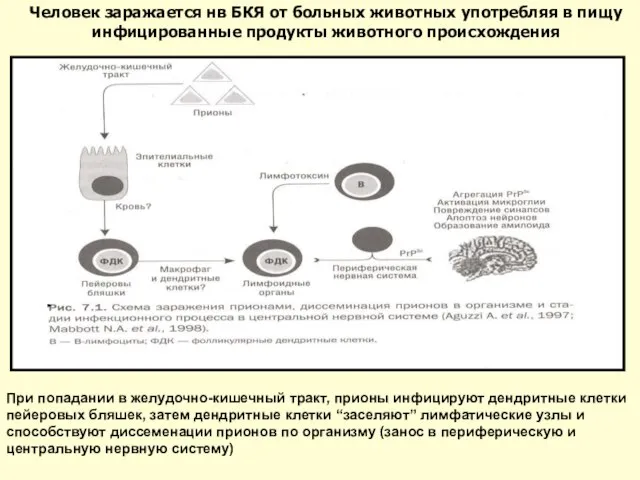
Слайд 38 В настоящее время полностью установлена связь между
нв БКЯ и ГЭК,
В настоящее время полностью установлена связь между
нв БКЯ и ГЭК,

Слайд 39По данным CDC (США) с момента регистрации первого случая нового варианта болезни
По данным CDC (США) с момента регистрации первого случая нового варианта болезни

Слайд 40Ятрогенная БКЯ
Ятрогенная БКЯ может рассматривается как внутрибольничная инфекция.
Все выявленные случаи
Ятрогенная БКЯ
Ятрогенная БКЯ может рассматривается как внутрибольничная инфекция.
Все выявленные случаи

Слайд 41
Гормон человеческого роста из гипофиза трупов применялся для лечения детей от
Гормон человеческого роста из гипофиза трупов применялся для лечения детей от

Слайд 42Первый случай в мире был зарегистрирован как “возможная БКЯ” у реципиента в
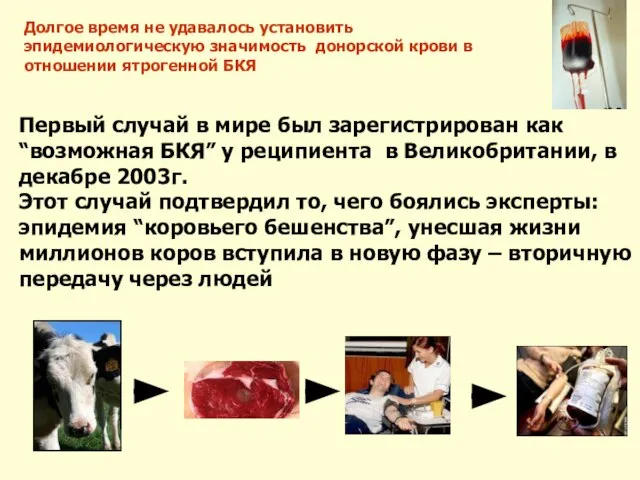
Слайд 43Случай 1.
Пациент – мужчина 62-х лет получил переливание крови в 1997г при
Случай 1.
Пациент – мужчина 62-х лет получил переливание крови в 1997г при
Слайд 44В июле 2004г английское правительство подтвердило наличие второго в Великобритании случая БКЯ
В июле 2004г английское правительство подтвердило наличие второго в Великобритании случая БКЯ

Слайд 45Последний (третий) случай заболевания зарегистрирован в декабре 2006г у молодого человека, 32-х
Последний (третий) случай заболевания зарегистрирован в декабре 2006г у молодого человека, 32-х

Слайд 46По результатам опубликованного в 2006г эпидемиологического исследования (Hewitt PE, 2006г)
24 пациента
По результатам опубликованного в 2006г эпидемиологического исследования (Hewitt PE, 2006г)
24 пациента

Слайд 47При заражении путем переливания крови регистрируется более короткий инкубационный период болезни, чем
При заражении путем переливания крови регистрируется более короткий инкубационный период болезни, чем

Слайд 48Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, Andrews N, Hilton
Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, Andrews N, Hilton

Слайд 49В 2004г в Великобритании было принято
решение исключить из донорства всех
В 2004г в Великобритании было принято
решение исключить из донорства всех

Слайд 50Донорской кровью, заготовленной в Великобритании, пользовались 11 стран мира, и исключить уже
Донорской кровью, заготовленной в Великобритании, пользовались 11 стран мира, и исключить уже

Слайд 51Великобритания импортировала в 2005г кровь из США на сумму 116 миллионов долларов
Великобритания импортировала в 2005г кровь из США на сумму 116 миллионов долларов

Слайд 52В связи с необходимостью прижизненной диагностики прионных болезней у лиц, находящихся в
В связи с необходимостью прижизненной диагностики прионных болезней у лиц, находящихся в

Слайд 53Дифференциальный диагноз болезни Крейтцфельдта – Якоба
проводится со всеми болезнями, одним из
Дифференциальный диагноз болезни Крейтцфельдта – Якоба
проводится со всеми болезнями, одним из

Слайд 54Биологические методы диагностики
Трансгенные мыши , несущие ген, кодирующий нормальный PrP человека
Биологические методы диагностики
Трансгенные мыши , несущие ген, кодирующий нормальный PrP человека

Слайд 55 Профилактика:
Исключение способов проникновения возбудителя в организм
(ограничено применение препаратов,
Профилактика:
Исключение способов проникновения возбудителя в организм
(ограничено применение препаратов,

Слайд 56ВОЗ, на современном этапе рекомендует 3 типа обработки не одноразового медицинского инструментария:
ВОЗ, на современном этапе рекомендует 3 типа обработки не одноразового медицинского инструментария:

 Единая информационная база домашних животных и животных без владельцев
Единая информационная база домашних животных и животных без владельцев Архиваторы.
Архиваторы. Условия обеспечения финансовой устойчивости страховщиков
Условия обеспечения финансовой устойчивости страховщиков Откуда берутся снежинки?
Откуда берутся снежинки? Рисковая модель оценки достаточности капитала (платежеспособности) страховщика в принципах и рекомендациях Международной ассоц
Рисковая модель оценки достаточности капитала (платежеспособности) страховщика в принципах и рекомендациях Международной ассоц Цветы в вазе. Натюрморт
Цветы в вазе. Натюрморт Capital punishment
Capital punishment  Виды кабелей
Виды кабелей Совместная проектная деятельность обучающихся как инструмент творческой интеграции
Совместная проектная деятельность обучающихся как инструмент творческой интеграции Геоинформационная система с веб-интерфейсом для поиска коммерческих и домашних точек питания
Геоинформационная система с веб-интерфейсом для поиска коммерческих и домашних точек питания Порядок учета средств криптографической защиты информации
Порядок учета средств криптографической защиты информации Управління персоналом та якістю проекту
Управління персоналом та якістю проекту Утомление при физической и умственной работе
Утомление при физической и умственной работе Информация и информационные процессы в живой и неживой природе
Информация и информационные процессы в живой и неживой природе Презентация на тему Древнейшие цивилизации
Презентация на тему Древнейшие цивилизации Банковская система
Банковская система Как снимается кинофильм
Как снимается кинофильм Презентация на тему гражданин российской федерации 11 класс
Презентация на тему гражданин российской федерации 11 класс Нестандартные приемы решения тригонометрических задач
Нестандартные приемы решения тригонометрических задач Стимулирование мыслительной деятельности учащихся через использование активных методов и приёмов обучения на уроках истории
Стимулирование мыслительной деятельности учащихся через использование активных методов и приёмов обучения на уроках истории Знающий тайну шахмат - знает тайну жизни
Знающий тайну шахмат - знает тайну жизни CATIA Организация связей
CATIA Организация связей Конфликты в школе
Конфликты в школе Абдоминальный болевой синдром
Абдоминальный болевой синдром  Презентация на тему Изобретения 19 века
Презентация на тему Изобретения 19 века У синих скал
У синих скал Презентация на тему Социальная информатика
Презентация на тему Социальная информатика  Волновые явления
Волновые явления