- ВАЛИДАЦИЯ

Содержание
- 4. До начала работ по валидации процесса необходимо завершить квалификацию критического оборудования и вспомогательных систем. Квалификацию обычно
- 6. Технологический регламент – это нормативный документ, в котором изложены технологические методы, технические средства, нормы и нормативы
- 7. Руководство по валидации процесса производства лекарственных препаратов для медицинского применения. Утверждено 26 сентября 2017 (вступает в
- 8. По определению PIC/S, валидация это: «Действия, которые в соответствии с принципами GMP доказывают, что определенная методика,
- 9. ВАЛИДАЦИЯ ПРОЦЕССА Валидация процесса производства является документированным подтверждением того, что процесс в пределах установленных параметров может
- 10. процедура, дающая высокую степень уверенности в том, что конкретный процесс, метод или система будет последовательно приводить
- 11. Валидация необходима при: внедрении новых средств производства, контроля качества, систем обеспечения производства; внедрении новых лекарственных средств;
- 12. Общие положения Независимо от используемого при разработке лекарственного препарата подхода, традиционного или расширенного, до начала реализации
- 13. Валидация сама по себе не улучшает качества продукции. Ее результаты могут либо повысить степень гарантии качества,
- 14. Результаты валидации оформляются Отчетом о проведении валидации. Отчет оформляется отдельно для каждого конкретного вида продукта. Валидации
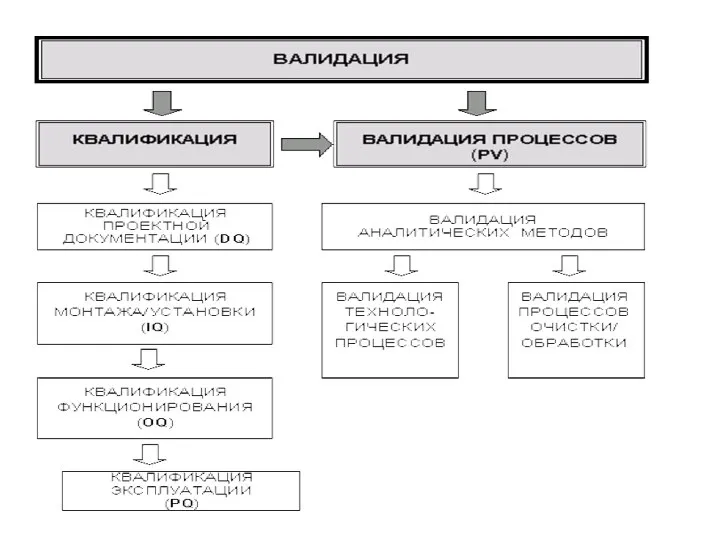
- 15. Квалификация (Qualification). Начальный этап валидации, который проводится для проверки и оценки проектной документации и условий производства
- 16. а) иногда работы по квалификации на стадиях OQ и PQ возможно и целесообразно проводить одновременно (например,
- 17. . Этапы валидации: 1. Квалификация (Qualification). 2. Валидация процессов (Process Validation - PV). Результаты всех стадий
- 18. . Виды валидации: Перспективная валидация. Проводится на вновь вводимом или реконструируемом производстве перед его пуском. При
- 19. Валидация оборудования Все производители оборудования указывают набор определенных характеристик своего продукта. Сюда относят: требуемые условия эксплуатации,
- 20. Валидация процесса Традиционная валидация процесса Традиционная валидация процесса, как правило, выполняется по завершении фармацевтической разработки и
- 21. Проведение полных валидационных исследований на опытно-промышленных сериях, в целом, считается нецелесообразным, поэтому для каждого лекарственного препарата
- 22. Должны быть представлены данные по валидации как минимум трех серий промышленного масштаба, если не обосновано иное
- 23. Для внедрения и поддержания непрерывной верификации процесса требуются знание и понимание процесса. Масштаб и степень применения
- 24. Непрерывная верификация процесса может быть введена на любом этапе жизненного цикла продукта. Этот подход может быть
- 25. Валидация процесса начинается уже на стадии его разработки. Полностью процесс валидируется при внедрении его в коммерческое
- 26. Разработка фармацевтического продукта и процесса его производства На первой стадии жизненного цикла производится сбор данных о
- 27. Технологическое пространство процесса содержит, с одной стороны, требования ко всем элементам технологического процесса, т.е. к сырью
- 28. Рекомендуется применять и различные инструменты анализа рисков, в частности, систему НАССР (Hazard Analysis and Critical Control
- 29. Квалификация эксплуатации (Performance Qualification – PQ). PQ- является вторым элементом стадии квалификации технологического процесса и проводится
- 30. Успешное выполнение PQ, показывает, что достигнут важный пункт в жизненном цикле продукции. Т.к. именно успешное выполнение
- 31. Протокол квалификация эксплуатации (Protocol PQ). Для этой стадии валидации процесса необходимо наличие письменного документа (протокола), который
- 32. Комбинированный подход Допускается использование также комбинированного подхода, заключающегося в применении традиционного подхода к валидации и непрерывной
- 33. Если не было показано что параметры, изученные при разработке проектного поля, масштабируются независимо от масштаба производства,
- 34. Масштабирование Во избежание повторения длительных и дорогостоящих испытаний необходимо должным образом организовать сбор информации и данных
- 35. Пострегистрационный контроль изменений Необходимо установить четкие процедуры для управления изменениями, предлагаемыми для производственного процесса. Такие процедуры
- 36. Общая схема проведения валидации на действующем производстве
- 37. Для планирования валидации используется документация 1. Проектная документация, разработанная в установленном порядке. 2. Приемно-сдаточная документация, подтверждающая
- 38. Примерное содержание валидационного плана (Рекомендуемое) Валидационный план включает в себя следующие положения, информацию, документы: 1. Цели
- 39. Схема валидации процесса в регистрационном досье должна содержать следующую информацию: краткое описание процесса с указанием критических
- 42. Скачать презентацию
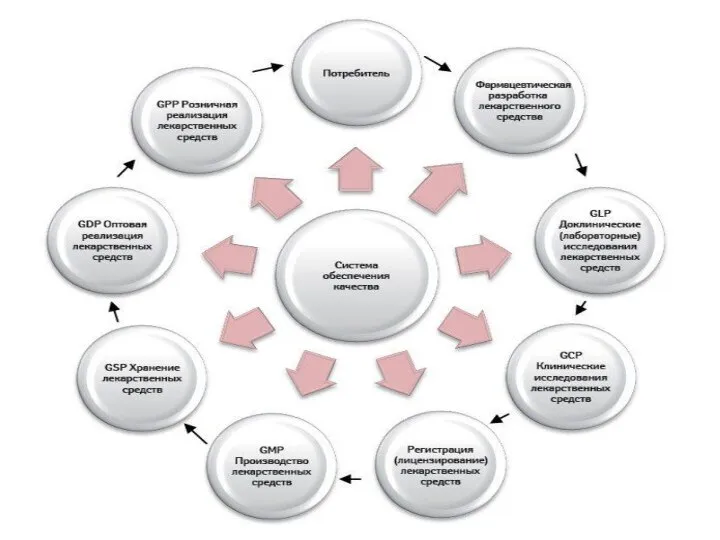
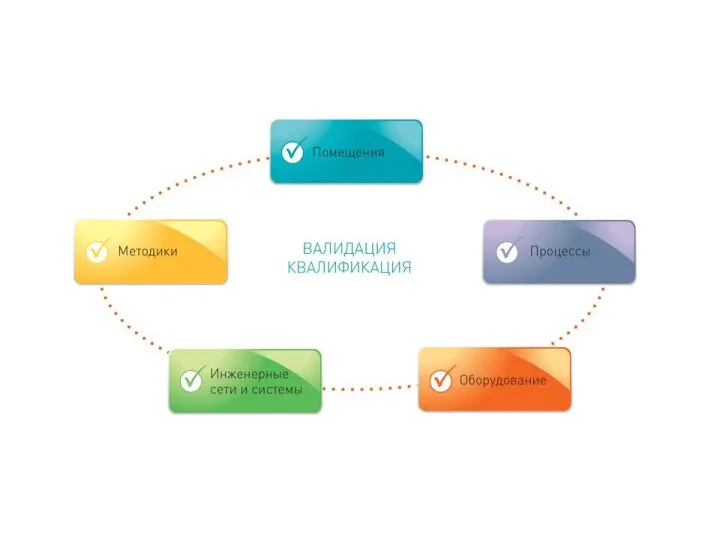
Слайд 4До начала работ по валидации процесса необходимо завершить квалификацию критического оборудования и
До начала работ по валидации процесса необходимо завершить квалификацию критического оборудования и

Слайд 6Технологический регламент – это нормативный документ, в котором изложены технологические методы, технические
Технологический регламент – это нормативный документ, в котором изложены технологические методы, технические

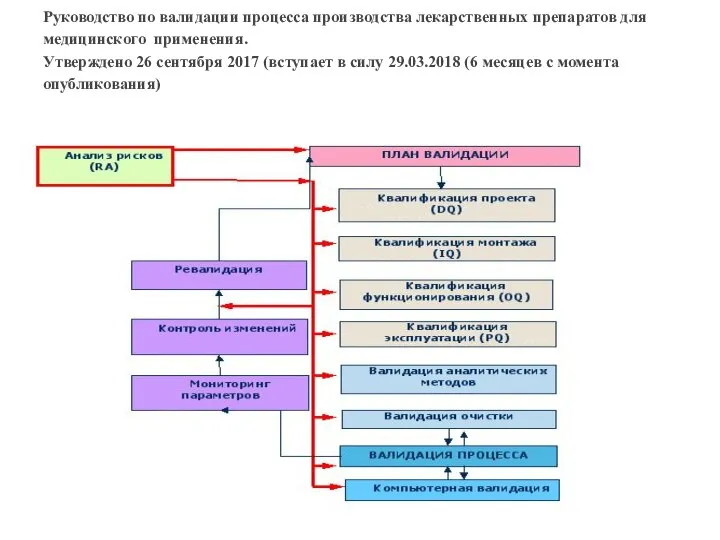
Слайд 7Руководство по валидации процесса производства лекарственных препаратов для медицинского применения.
Утверждено 26 сентября
Руководство по валидации процесса производства лекарственных препаратов для медицинского применения. Утверждено 26 сентября
Слайд 8По определению PIC/S, валидация это:
«Действия, которые в соответствии с принципами GMP доказывают,
По определению PIC/S, валидация это: «Действия, которые в соответствии с принципами GMP доказывают,

Слайд 9ВАЛИДАЦИЯ ПРОЦЕССА
Валидация процесса производства является документированным подтверждением того, что процесс в пределах
ВАЛИДАЦИЯ ПРОЦЕССА
Валидация процесса производства является документированным подтверждением того, что процесс в пределах

Слайд 10процедура, дающая высокую степень уверенности в том, что конкретный процесс, метод или
процедура, дающая высокую степень уверенности в том, что конкретный процесс, метод или

Слайд 11Валидация необходима при:
внедрении новых средств производства, контроля качества, систем обеспечения производства;
Валидация необходима при:
внедрении новых средств производства, контроля качества, систем обеспечения производства;

Слайд 12Общие положения
Независимо от используемого при разработке лекарственного препарата подхода, традиционного или
Общие положения
Независимо от используемого при разработке лекарственного препарата подхода, традиционного или

Слайд 13Валидация сама по себе не улучшает качества продукции. Ее результаты могут либо
Валидация сама по себе не улучшает качества продукции. Ее результаты могут либо

Слайд 14Результаты валидации оформляются Отчетом о проведении валидации. Отчет оформляется отдельно для каждого
Результаты валидации оформляются Отчетом о проведении валидации. Отчет оформляется отдельно для каждого

Слайд 15Квалификация (Qualification).
Начальный этап валидации, который проводится для проверки и оценки проектной
Квалификация (Qualification).
Начальный этап валидации, который проводится для проверки и оценки проектной

Слайд 16а) иногда работы по квалификации на стадиях OQ и PQ возможно и
а) иногда работы по квалификации на стадиях OQ и PQ возможно и

Слайд 17. Этапы валидации:
1. Квалификация (Qualification).
2. Валидация процессов (Process Validation -
. Этапы валидации:
1. Квалификация (Qualification).
2. Валидация процессов (Process Validation -

Слайд 18. Виды валидации:
Перспективная валидация. Проводится на вновь вводимом или реконструируемом производстве
. Виды валидации:
Перспективная валидация. Проводится на вновь вводимом или реконструируемом производстве

Слайд 19Валидация оборудования
Все производители оборудования указывают набор определенных характеристик своего продукта.
Сюда относят:
Валидация оборудования
Все производители оборудования указывают набор определенных характеристик своего продукта.
Сюда относят:

Слайд 20Валидация процесса
Традиционная валидация процесса
Традиционная валидация процесса, как правило, выполняется по завершении
Валидация процесса
Традиционная валидация процесса
Традиционная валидация процесса, как правило, выполняется по завершении

Слайд 21Проведение полных валидационных исследований на опытно-промышленных сериях, в целом, считается нецелесообразным, поэтому
Проведение полных валидационных исследований на опытно-промышленных сериях, в целом, считается нецелесообразным, поэтому

Слайд 22Должны быть представлены данные по валидации как минимум трех серий промышленного масштаба,
Должны быть представлены данные по валидации как минимум трех серий промышленного масштаба,

Слайд 23Для внедрения и поддержания непрерывной верификации процесса требуются знание и понимание процесса.
Для внедрения и поддержания непрерывной верификации процесса требуются знание и понимание процесса.

Слайд 24Непрерывная верификация процесса может быть введена на любом этапе жизненного цикла продукта.
Непрерывная верификация процесса может быть введена на любом этапе жизненного цикла продукта.

Слайд 25Валидация процесса начинается уже на стадии его разработки. Полностью процесс валидируется при
Валидация процесса начинается уже на стадии его разработки. Полностью процесс валидируется при

Слайд 26Разработка фармацевтического продукта и процесса его производства
На первой стадии жизненного цикла
Разработка фармацевтического продукта и процесса его производства
На первой стадии жизненного цикла

Слайд 27Технологическое пространство процесса содержит, с одной стороны, требования ко всем элементам технологического
Технологическое пространство процесса содержит, с одной стороны, требования ко всем элементам технологического

Слайд 28Рекомендуется применять и различные инструменты анализа рисков, в частности, систему НАССР (Hazard
Рекомендуется применять и различные инструменты анализа рисков, в частности, систему НАССР (Hazard

Слайд 29Квалификация эксплуатации
(Performance Qualification – PQ).
PQ- является вторым элементом стадии квалификации
Квалификация эксплуатации
(Performance Qualification – PQ).
PQ- является вторым элементом стадии квалификации

Слайд 30Успешное выполнение PQ, показывает, что достигнут важный пункт в жизненном цикле продукции.
Успешное выполнение PQ, показывает, что достигнут важный пункт в жизненном цикле продукции.

Слайд 31Протокол квалификация эксплуатации (Protocol PQ).
Для этой стадии валидации процесса необходимо наличие письменного
Протокол квалификация эксплуатации (Protocol PQ).
Для этой стадии валидации процесса необходимо наличие письменного

Слайд 32Комбинированный подход
Допускается использование также комбинированного подхода, заключающегося в применении традиционного подхода
Комбинированный подход
Допускается использование также комбинированного подхода, заключающегося в применении традиционного подхода

Слайд 33Если не было показано что параметры, изученные при разработке проектного поля, масштабируются
Если не было показано что параметры, изученные при разработке проектного поля, масштабируются

Слайд 34Масштабирование
Во избежание повторения длительных и дорогостоящих испытаний необходимо должным образом организовать
Масштабирование
Во избежание повторения длительных и дорогостоящих испытаний необходимо должным образом организовать

Слайд 35Пострегистрационный контроль изменений
Необходимо установить четкие процедуры для управления изменениями, предлагаемыми для
Пострегистрационный контроль изменений
Необходимо установить четкие процедуры для управления изменениями, предлагаемыми для

Слайд 36Общая схема проведения валидации на действующем производстве
Общая схема проведения валидации на действующем производстве

Слайд 37Для планирования валидации используется документация
1. Проектная документация, разработанная в установленном порядке.
2.
Для планирования валидации используется документация
1. Проектная документация, разработанная в установленном порядке. 2.

Слайд 38Примерное содержание валидационного плана
(Рекомендуемое)
Валидационный план включает в себя следующие положения, информацию,
Примерное содержание валидационного плана
(Рекомендуемое)
Валидационный план включает в себя следующие положения, информацию,

Слайд 39Схема валидации процесса в регистрационном досье должна
содержать следующую информацию:
краткое описание процесса
Схема валидации процесса в регистрационном досье должна
содержать следующую информацию:
краткое описание процесса

 Презентация на тему Альтернативные источники электроэнергии
Презентация на тему Альтернативные источники электроэнергии А) Существительное, прилагательное, наречие, глагол.Б) Чисто, красивый, быстро, прекрасноВ) Свежо, горячо, жгуче, хорошо.Г) Издалека,
А) Существительное, прилагательное, наречие, глагол.Б) Чисто, красивый, быстро, прекрасноВ) Свежо, горячо, жгуче, хорошо.Г) Издалека, За страницами школьного учебника
За страницами школьного учебника Курация больного
Курация больного  Тема: Работа с бумагой и картоном Аппликация. Объемная аппликация.«Коровка»
Тема: Работа с бумагой и картоном Аппликация. Объемная аппликация.«Коровка» Сборочный чертеж и спецификация
Сборочный чертеж и спецификация Дизайн-проект Спальня
Дизайн-проект Спальня Проектирование и архитектура вычислительных систем
Проектирование и архитектура вычислительных систем Украинская Umbro Лига
Украинская Umbro Лига Презентация на тему Планирование родительского собрания
Презентация на тему Планирование родительского собрания Хозяйственное освоение Сибири
Хозяйственное освоение Сибири Где логика?, универсиада, виды спорта
Где логика?, универсиада, виды спорта Презентация на тему Пчелы и геометрия
Презентация на тему Пчелы и геометрия  ПАДЕЖИ. Знакомство с падежами имен существительных
ПАДЕЖИ. Знакомство с падежами имен существительных Мы - россияне
Мы - россияне Інжу слайд 2
Інжу слайд 2 Сезоны и погода
Сезоны и погода Презентация на тему Русская культура
Презентация на тему Русская культура Огнестрельная травма
Огнестрельная травма ПРЕЗЕНТАЦИЯ 213-КАРАР 26.09.22 КК
ПРЕЗЕНТАЦИЯ 213-КАРАР 26.09.22 КК ФОРМИРОВАНИЕ ЦЕЛЕЙ УПРАВЛЕНИЯ ПРОЦЕССАМИ В СИСТЕМАХ МЕНЕДЖМЕНТА
ФОРМИРОВАНИЕ ЦЕЛЕЙ УПРАВЛЕНИЯ ПРОЦЕССАМИ В СИСТЕМАХ МЕНЕДЖМЕНТА Презентация на тему Лучший ученик
Презентация на тему Лучший ученик Применение технологий, методов и средств обучения.
Применение технологий, методов и средств обучения. Электронные таблицы
Электронные таблицы Внешний контроль и надзор за законностью в сфере деятельности исполнительной власти
Внешний контроль и надзор за законностью в сфере деятельности исполнительной власти Относимость и допустимость доказательств
Относимость и допустимость доказательств Основные показатели надёжности
Основные показатели надёжности Отчет о работе Управляющего Совета
Отчет о работе Управляющего Совета