- Заболевания нервно-мышечной системы

Содержание
- 2. ОПРЕДЕЛЕНИЕ Нервно-мышечные заболевания – болезни с поражением нейронов, их аксонов, синапсов или самих мышц Болезнь мышечной
- 3. ПРИЗНАКИ ПОРАЖЕНИЯ Гипотония Гипорефлексия Гипотрофия
- 4. КРУГ НЕРВНО-МЫШЕЧНЫХ ЗАБОЛЕВАНИЙ Наследственные заболевания Инфекционные Воспалительные Паранеопластические При соматических заболеваниях При эндокринных заболеваниях и др.
- 5. НЕРВНО-МЫШЕЧНАЯ ЕДИНИЦА А Б В Г
- 6. КЛАССИФИКАЦИЯ А Заболевания, связанные с поражением передних рогов спинного мозга Б Болезни, связанные с поражением периферических
- 7. ПОРАЖЕНИЕ ПЕРЕДНЕГО РОГА Только двигательные нарушения + фасцикуляции Отсутствуют чувствительные нарушения
- 8. ПОРАЖЕНИЕ ПЕРИФЕРИЧЕСКИХ НЕРВОВ Дистальные парезы (кисти, стопы) В подавляющем большинстве случаев + расстройства чувствительности + вегетативные
- 9. БОЛЕЗНИ СИНАПСА Патологическая мышечная утомляемость
- 10. МЫШЕЧНОЕ ПОРАЖЕНИЕ Преимущественное поражение проксимальных отделов конечностей (тазовый, плечевой пояс) Отсутствуют чувствительные нарушения
- 11. ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ ЭМГ Глобальная (поверхностные электроды, суммарная активность) Игольчатая (активность отдельного мышечного волокна) Скорость проведения возбуждения
- 12. ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ Исследование ферментов крови ↑ КФК - креатинфосфокиназа ↑ ЛДГ – лактатдегидрогеназа Исследование электролитов крови
- 13. ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ Биопсия мышц (гистохимическое исследование) Составление родословных таблиц
- 14. БОЛЕЗНИ ПЕРЕДНЕГО РОГА Инфекционные Клещевой энцефалит Полиомиелит (болезнь Гейне-Медина)- эпидемический детский паралич вирусной этиологии Полиомиелитоподобные заболевания
- 15. БОЛЕЗНИ ПЕРЕДНЕГО РОГА Дегенеративные заболевания Боковой амиотрофический склероз (БАС) – болезнь Шарко дегенерация боковых столбов поражение
- 16. БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ Пирамидный путь Передний рог
- 17. УРОВНИ ПОРАЖЕНИЯ ПРИ БАС Шейное утолщение Поясничное утолщение Бульбарный отдел ствола головного мозга
- 18. КЛИНИКА БАС Смешанные парезы в руках и/или ногах: признаки вовлечения переднего рога (фасцикуляции, атрофии) признаки поражения
- 19. ТЕЧЕНИЕ БАС Течение хроническое или подострое Варианты течения: как правило, восходящее, но может быть начало с
- 20. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ - СПИНАЛЬНЫЕ АМИОТРОФИИ Ранняя детская форма Вернига-Гофманна Юношеская форма Кугельберга-Веландер Бульбо-спинальная амиотрофия Кеннеди
- 21. РАННЯЯ ДЕТСКАЯ ФОРМА ВЕРНИГА - ГОФМАННА Описана в 1891 году Острая злокачественная инфантильная спинальная амиотрофия Тип
- 22. КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА Период внутриутробного развития – отсутствие или слабое шевеление плода у 1/3 матерей
- 23. КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА «вялый ребенок»
- 24. КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА Мышечная гипотония
- 25. ЮНОШЕСКАЯ ФОРМА КУГЕЛЬБЕРГА - ВЕЛАНДЕР Описана в 1956 году Течение доброкачественное Тип наследования – аутосомно-рецессивный
- 26. КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ КУГЕЛЬБЕРГА-ВЕЛАНДЕР Начало заболевания в 2 -15 лет (в среднем в 5 лет) Очень
- 27. СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ Тяжелая атрофия мышц плечевого пояса
- 28. БУЛЬБО-СПИНАЛЬНАЯ АМИОТРОФИЯ КЕННЕДИ Описана в 1968 году Доброкачественное течение Тип наследования – рецессивный, сцепленный с Х-хромосомой
- 29. СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ Мать-носитель Здоровый отец Здоровый мужчина Здоровая женщина Больной мужчина Женщина-носитель Хх
- 30. КЛИНИКА БУЛЬБО-СПИНАЛЬНОЙ АМИОТРОФИИ КЕННЕДИ Начало в зрелом возрасте (4-я декада) Проксимальная слабость в руках, ногах Бульбарный
- 31. БОЛЕЗНИ, СВЯЗАННЫЕ С ПОРАЖЕНИЕМ ПЕРИФЕРИЧЕСКИХ НЕРВОВ Общие признаки: периферические парезы полиневритический тип нарушения чувствительности вегетативные нарушения
- 32. ВИДЫ ПОЛИНЕЙРОПАТИЙ Аксональные полинейропатии: при дефиците тиамина, рибофлавина при отравлении мышьяком лекарственные полинейропатии (нитрофуран, изониазид, пиридоксин
- 33. НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ Группа наследственных сенсомоторных полинейропатий Описана Ж.Шарко, П.Мари в 1886 г. и Г.Тус в
- 34. КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ Начало с ног: постепенно нарастает слабость и атрофии мышц голеней, мелких мышц
- 35. КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ А Б А – дистальные атрофии Б – полиневритический тип нарушения чувствительности
- 36. Атрофия мышц кисти, «когтистая лапа» Невральная амиотрофия Шарко-Мари
- 37. Стопа Фридрайха, «полая» стопа Невральная амиотрофия Шарко-Мари
- 38. НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ Типы наследования: Аутосомно-доминантный Аутосомно-рецессивный Сцепленный с Х-хромосомой
- 39. ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА Синдром патологической мышечной утомляемости – нарастание пареза (слабости) к вечеру
- 40. ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА Ботулизм Миастения Миастенические синдромы
- 41. ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА Ботулизм возникает преходящая блокада пресинаптических структур, нарушается холинергическая трансмиссия связан с воздействием токсина
- 42. ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА Миастения (Myasthenia gravis) Заболевание аутоиммунной природы Вырабатываются антитела к белку ацетилхолиновых рецепторов (есть
- 43. КЛИНИКА МИАСТЕНИИ Начало, как правило, с глазных мышц (птоз, двоение, сонное выражение лица) Часто начало с
- 44. КЛИНИЧЕСКИЕ ПРОБЫ, ВЫЯВЛЯЮЩИЕ ПАТОЛОГИЧЕСКУЮ МЫШЕЧНУЮ УТОМЛЯЕМОСТЬ Фиксировать взор вверх – 30 секунд (больной устает) Громко считать
- 45. ТЕЧЕНИЕ МИАСТЕНИИ Характерно течение с ремиссиями В 20% наблюдаются миастенические кризы (генерализованная мышечная слабость, бульбарные, дыхательные
- 46. ЛЕЧЕНИЕ МИАСТЕНИИ Антихолинэстеразные прозерин – быстрого действия калимин – медленного Преднизолон (больным старше 50 лет) Плазмаферез
- 47. МИАСТЕНИЧЕСКИЕ СИНДРОМЫ Синдром Ламберта-Итона – паранеопластический синдром Наблюдается у мужчин старше 40 лет при бронхогенном раке
- 48. МИАСТЕНИЧЕСКИЕ СИНДРОМЫ Клиника не страдают глазные мышцы нет реакции на антихолинэстеразные препараты миастенический синдром может опережать
- 49. МИАСТЕНИЧЕСКИЕ СИНДРОМЫ Другие причины Заболевания щитовидной железы (аутоиммунные) Интоксикация лекарственными препаратами: неомицин гентамицин Д-пенициламин (при лечении
- 50. БОЛЕЗНИ, СВЯЗАННЫЕ С ПОРАЖЕНИЕМ МЫШЦ (МИОПАТИИ) Общие признаки: Отсутствие чувствительных и вегетативных нарушений Поражение проксимальных отделов
- 51. ПОЛИМИОЗИТ Не наследственное (аутоиммунное) заболевание Нарушение клеточного и гуморального иммунитета («воспалительная миопатия») Может протекать с кожными
- 52. КЛИНИКА ПОЛИМИОЗИТА Начало – острое или подострое Мышечная слабость Недомогание, артралгии, миалгии Повышение температуры тела Поражение
- 53. ПОЛИМИОЗИТ Данные дополнительного исследования: Уровень КФК не коррелирует с тяжестью ↑ миоглобин в сыворотке крови ↑СОЭ
- 54. ВОСПАЛИТЕЛЬНАЯ МИОПАТИЯ (ПОЛИМИОЗИТ) I Биопсия мышцы Воспалительные изменения в мышце
- 55. НАСЛЕДСТВЕННЫЕ МИОДИСТРОФИИ (МИОПАТИИ) Известно много форм и вариантов Псевдогипертрофическая миодистрофия Дюшена Миодистрофия Ландузи-Дежерина Миодистрофия Эрба
- 56. Неравномерность диаметра мышечных волокон, разрастание соединительной и жировой тканей ПЕРВИЧНЫЕ АМИОТРОФИИ (МИОПАТИИ) гистологическая картина четырехглавой мышцы
- 57. ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МИОДИСТРОФИЯ ДЮШЕНА Описана в 1861 году Дюшеном В 1879 году Говерс обобщил материал: 21 больной
- 58. КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА Первые признаки появляются с момента начала ходьбы (близко колени, ноги ставятся на
- 59. КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА Умственная отсталость – 30%. ЭМГ – признаки первично-мышечного поражения. Биопсия мышц –
- 60. КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА псевдогипертрофии
- 61. Мальчик 5 лет Наблюдаются псевдогипертрофии мышц, лордоз ПСЕВДОГИПЕРТРОФИЧЕСКАЯ ФОРМА ДЮШЕНА
- 62. СИМПТОМ «ВСТАВАНИЯ ЛЕСЕНКОЙ» ПРИ МИОПАТИИ
- 63. МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА Описана в 1884 -1886 г.г. (Дежерин, Ландузи) Частота встречаемости–0,4 на 100 000 населения Тип
- 64. КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА Дебют в 15-25 лет (≈20 лет) Слабость и атрофии мышц лица, плечевого пояса,
- 65. КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА Слабость мышц лица: глазные щели не смыкаются ночью больные не могут свистеть, пить
- 66. КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА Атрофия и слабость мышц плечевого и тазового поясов: крыловидные лопатки, гипотрофия передней лестничной
- 67. КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА Интеллект не страдает КФК↑ у 50 - 80% ЛДГ↑ у 20% Альдолаза↑ у
- 68. ПЛЕЧЕЛОПАТОЧНО-ЛИЦЕВАЯ МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА Поражение мышц лица и плечевого пояса Мальчик 13 лет, болен с 7 лет,
- 69. ЛОПАТОЧНО-ПЕРОНЕАЛЬНАЯ ФОРМА Выражена атрофия мышц плечевого пояса и перонеальных мышц Мальчик 15 лет, болен с 7
- 70. МИОДИСТРОФИЯ ЭРБА-РОТА Описана в 1884 году Эрбом Тип наследования аутосомно-рецессивный Экспрессивность гена у членов семьи разная
- 71. КЛИНИКА МИОДИСТРОФИИ ЭРБА Дебют во 2-м десятилетии, но может быть и в детстве и после 30
- 72. КЛИНИКА МИОДИСТРОФИИ ЭРБА Форма Лейдена-Мебиуса начало с проксимальных отделов ног Форма Эрба начало с плечевого пояса+спина,
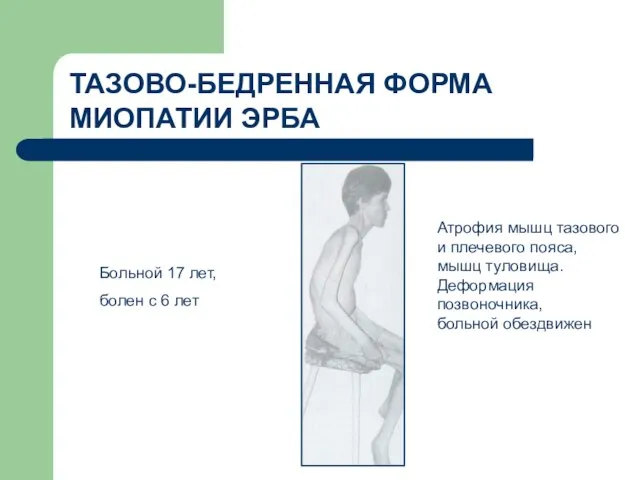
- 73. ТАЗОВО-БЕДРЕННАЯ ФОРМА МИОПАТИИ ЭРБА Больной 17 лет, болен с 6 лет Атрофия мышц тазового и плечевого
- 75. Скачать презентацию
Слайд 2ОПРЕДЕЛЕНИЕ
Нервно-мышечные заболевания – болезни с поражением нейронов, их аксонов, синапсов или самих
ОПРЕДЕЛЕНИЕ
Нервно-мышечные заболевания – болезни с поражением нейронов, их аксонов, синапсов или самих

Слайд 3ПРИЗНАКИ ПОРАЖЕНИЯ
Гипотония
Гипорефлексия
Гипотрофия
ПРИЗНАКИ ПОРАЖЕНИЯ
Гипотония
Гипорефлексия
Гипотрофия

Слайд 4КРУГ НЕРВНО-МЫШЕЧНЫХ ЗАБОЛЕВАНИЙ
Наследственные заболевания
Инфекционные
Воспалительные
Паранеопластические
При соматических заболеваниях
При эндокринных заболеваниях и др.
КРУГ НЕРВНО-МЫШЕЧНЫХ ЗАБОЛЕВАНИЙ
Наследственные заболевания
Инфекционные
Воспалительные
Паранеопластические
При соматических заболеваниях
При эндокринных заболеваниях и др.

Слайд 5НЕРВНО-МЫШЕЧНАЯ ЕДИНИЦА
А
Б
В
Г
НЕРВНО-МЫШЕЧНАЯ ЕДИНИЦА
А
Б
В
Г
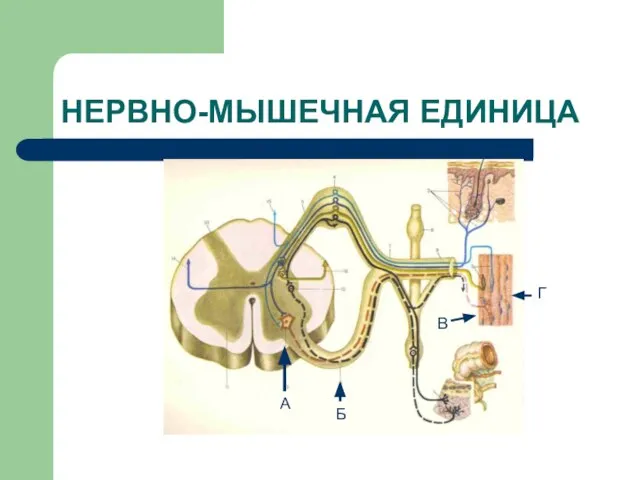
Слайд 6КЛАССИФИКАЦИЯ
А Заболевания, связанные с поражением
передних рогов спинного мозга
Б Болезни, связанные с
КЛАССИФИКАЦИЯ
А Заболевания, связанные с поражением
передних рогов спинного мозга
Б Болезни, связанные с

Слайд 7ПОРАЖЕНИЕ ПЕРЕДНЕГО РОГА
Только двигательные нарушения
+ фасцикуляции
Отсутствуют чувствительные нарушения
ПОРАЖЕНИЕ ПЕРЕДНЕГО РОГА
Только двигательные нарушения
+ фасцикуляции
Отсутствуют чувствительные нарушения

Слайд 8ПОРАЖЕНИЕ ПЕРИФЕРИЧЕСКИХ НЕРВОВ
Дистальные парезы (кисти, стопы)
В подавляющем большинстве случаев
+ расстройства чувствительности
+
ПОРАЖЕНИЕ ПЕРИФЕРИЧЕСКИХ НЕРВОВ
Дистальные парезы (кисти, стопы)
В подавляющем большинстве случаев
+ расстройства чувствительности
+

Слайд 9БОЛЕЗНИ СИНАПСА
Патологическая мышечная утомляемость
БОЛЕЗНИ СИНАПСА
Патологическая мышечная утомляемость

Слайд 10МЫШЕЧНОЕ ПОРАЖЕНИЕ
Преимущественное поражение проксимальных отделов конечностей (тазовый, плечевой пояс)
Отсутствуют чувствительные нарушения
МЫШЕЧНОЕ ПОРАЖЕНИЕ
Преимущественное поражение проксимальных отделов конечностей (тазовый, плечевой пояс)
Отсутствуют чувствительные нарушения

Слайд 11ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
ЭМГ
Глобальная
(поверхностные электроды, суммарная активность)
Игольчатая
(активность отдельного
ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
ЭМГ
Глобальная
(поверхностные электроды, суммарная активность)
Игольчатая
(активность отдельного

Слайд 12ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
Исследование ферментов крови
↑ КФК - креатинфосфокиназа
↑ ЛДГ – лактатдегидрогеназа
Исследование
ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
Исследование ферментов крови
↑ КФК - креатинфосфокиназа
↑ ЛДГ – лактатдегидрогеназа
Исследование

Слайд 13ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
Биопсия мышц (гистохимическое исследование)
Составление родословных таблиц
ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
Биопсия мышц (гистохимическое исследование)
Составление родословных таблиц

Слайд 14БОЛЕЗНИ ПЕРЕДНЕГО РОГА
Инфекционные
Клещевой энцефалит
Полиомиелит (болезнь Гейне-Медина)-
эпидемический детский паралич
БОЛЕЗНИ ПЕРЕДНЕГО РОГА
Инфекционные
Клещевой энцефалит
Полиомиелит (болезнь Гейне-Медина)-
эпидемический детский паралич

Слайд 15БОЛЕЗНИ ПЕРЕДНЕГО РОГА
Дегенеративные заболевания
Боковой амиотрофический склероз
(БАС) – болезнь Шарко
дегенерация боковых
БОЛЕЗНИ ПЕРЕДНЕГО РОГА
Дегенеративные заболевания
Боковой амиотрофический склероз
(БАС) – болезнь Шарко
дегенерация боковых

Слайд 16БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ
Пирамидный путь
Передний рог
БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ
Пирамидный путь
Передний рог
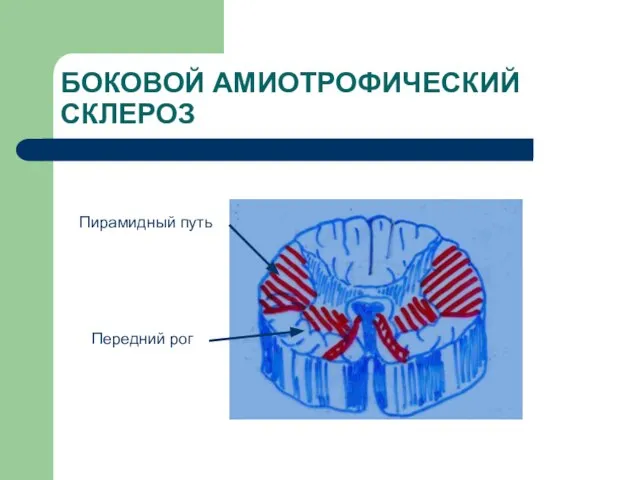
Слайд 17УРОВНИ ПОРАЖЕНИЯ ПРИ БАС
Шейное утолщение
Поясничное утолщение
Бульбарный отдел ствола головного мозга
УРОВНИ ПОРАЖЕНИЯ ПРИ БАС
Шейное утолщение
Поясничное утолщение
Бульбарный отдел ствола головного мозга

Слайд 18КЛИНИКА БАС
Смешанные парезы в руках и/или ногах:
признаки вовлечения переднего рога
(фасцикуляции,
КЛИНИКА БАС
Смешанные парезы в руках и/или ногах:
признаки вовлечения переднего рога
(фасцикуляции,

Слайд 19ТЕЧЕНИЕ БАС
Течение хроническое или подострое
Варианты течения:
как правило, восходящее, но может быть
ТЕЧЕНИЕ БАС
Течение хроническое или подострое
Варианты течения:
как правило, восходящее, но может быть

Слайд 20НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ - СПИНАЛЬНЫЕ АМИОТРОФИИ
Ранняя детская форма Вернига-Гофманна
Юношеская форма Кугельберга-Веландер
Бульбо-спинальная амиотрофия Кеннеди
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ - СПИНАЛЬНЫЕ АМИОТРОФИИ
Ранняя детская форма Вернига-Гофманна
Юношеская форма Кугельберга-Веландер
Бульбо-спинальная амиотрофия Кеннеди

Слайд 21РАННЯЯ ДЕТСКАЯ ФОРМА
ВЕРНИГА - ГОФМАННА
Описана в 1891 году
Острая злокачественная инфантильная спинальная
РАННЯЯ ДЕТСКАЯ ФОРМА
ВЕРНИГА - ГОФМАННА
Описана в 1891 году
Острая злокачественная инфантильная спинальная

Слайд 22КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
Период внутриутробного развития – отсутствие или слабое шевеление плода
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
Период внутриутробного развития – отсутствие или слабое шевеление плода

Слайд 23КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
«вялый ребенок»
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
«вялый ребенок»
Слайд 24КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
Мышечная гипотония
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
Мышечная гипотония

Слайд 25ЮНОШЕСКАЯ ФОРМА
КУГЕЛЬБЕРГА - ВЕЛАНДЕР
Описана в 1956 году
Течение доброкачественное
Тип наследования – аутосомно-рецессивный
ЮНОШЕСКАЯ ФОРМА
КУГЕЛЬБЕРГА - ВЕЛАНДЕР
Описана в 1956 году
Течение доброкачественное
Тип наследования – аутосомно-рецессивный

Слайд 26КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ КУГЕЛЬБЕРГА-ВЕЛАНДЕР
Начало заболевания в 2 -15 лет (в среднем в
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ КУГЕЛЬБЕРГА-ВЕЛАНДЕР
Начало заболевания в 2 -15 лет (в среднем в

Слайд 27
СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ
Тяжелая атрофия
мышц плечевого пояса
СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ
Тяжелая атрофия
мышц плечевого пояса

Слайд 28БУЛЬБО-СПИНАЛЬНАЯ АМИОТРОФИЯ КЕННЕДИ
Описана в 1968 году
Доброкачественное течение
Тип наследования – рецессивный, сцепленный с
БУЛЬБО-СПИНАЛЬНАЯ АМИОТРОФИЯ КЕННЕДИ
Описана в 1968 году
Доброкачественное течение
Тип наследования – рецессивный, сцепленный с

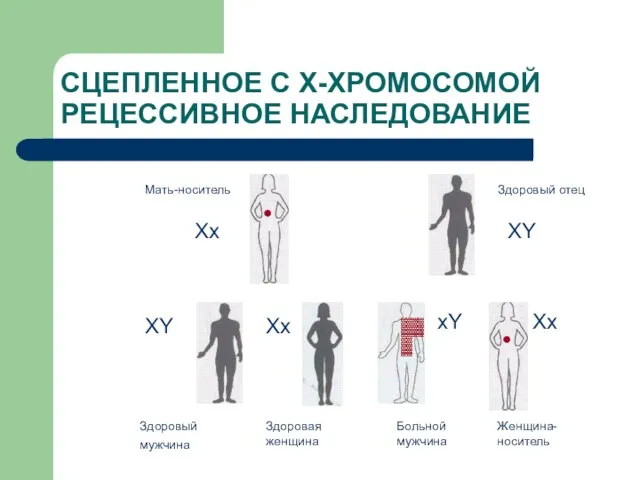
Слайд 29СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ
Мать-носитель
Здоровый отец
Здоровый мужчина
Здоровая женщина
Больной мужчина
Женщина-носитель
Хх
ХY
XY
Xx
xY
Xx
●
▓▓▓
●
СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ
Мать-носитель
Здоровый отец
Здоровый мужчина
Здоровая женщина
Больной мужчина
Женщина-носитель
Хх
ХY
XY
Xx
xY
Xx
●
▓▓▓
●
Слайд 30КЛИНИКА БУЛЬБО-СПИНАЛЬНОЙ АМИОТРОФИИ КЕННЕДИ
Начало в зрелом возрасте (4-я декада)
Проксимальная слабость в руках,
КЛИНИКА БУЛЬБО-СПИНАЛЬНОЙ АМИОТРОФИИ КЕННЕДИ
Начало в зрелом возрасте (4-я декада)
Проксимальная слабость в руках,

Слайд 31БОЛЕЗНИ, СВЯЗАННЫЕ
С ПОРАЖЕНИЕМ ПЕРИФЕРИЧЕСКИХ НЕРВОВ
Общие признаки:
периферические парезы
полиневритический тип нарушения
чувствительности
БОЛЕЗНИ, СВЯЗАННЫЕ
С ПОРАЖЕНИЕМ ПЕРИФЕРИЧЕСКИХ НЕРВОВ
Общие признаки:
периферические парезы
полиневритический тип нарушения
чувствительности

Слайд 32ВИДЫ ПОЛИНЕЙРОПАТИЙ
Аксональные полинейропатии:
при дефиците тиамина, рибофлавина
при отравлении мышьяком
лекарственные полинейропатии
ВИДЫ ПОЛИНЕЙРОПАТИЙ
Аксональные полинейропатии:
при дефиците тиамина, рибофлавина
при отравлении мышьяком
лекарственные полинейропатии

Слайд 33НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ
Группа наследственных сенсомоторных полинейропатий
Описана Ж.Шарко, П.Мари в 1886 г. и
НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ
Группа наследственных сенсомоторных полинейропатий
Описана Ж.Шарко, П.Мари в 1886 г. и

Слайд 34КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ
Начало с ног:
постепенно нарастает слабость и атрофии
КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ
Начало с ног:
постепенно нарастает слабость и атрофии

Слайд 35КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ
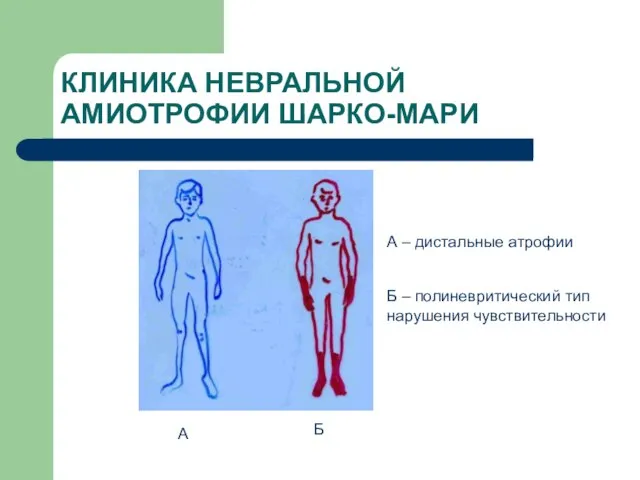
А
Б
А – дистальные атрофии
Б – полиневритический тип
КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ
А
Б
А – дистальные атрофии
Б – полиневритический тип
Слайд 36
Атрофия мышц кисти,
«когтистая лапа»
Невральная амиотрофия Шарко-Мари
Атрофия мышц кисти,
«когтистая лапа»
Невральная амиотрофия Шарко-Мари

Слайд 37
Стопа Фридрайха,
«полая» стопа
Невральная амиотрофия Шарко-Мари
Стопа Фридрайха,
«полая» стопа
Невральная амиотрофия Шарко-Мари

Слайд 38НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ
Типы наследования:
Аутосомно-доминантный
Аутосомно-рецессивный
Сцепленный с Х-хромосомой
НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ
Типы наследования:
Аутосомно-доминантный
Аутосомно-рецессивный
Сцепленный с Х-хромосомой

Слайд 39ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Синдром патологической мышечной утомляемости – нарастание пареза (слабости) к вечеру
ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Синдром патологической мышечной утомляемости – нарастание пареза (слабости) к вечеру

Слайд 40ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Ботулизм
Миастения
Миастенические синдромы
ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Ботулизм
Миастения
Миастенические синдромы

Слайд 41ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Ботулизм
возникает преходящая блокада пресинаптических структур, нарушается холинергическая трансмиссия
связан с воздействием
ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Ботулизм
возникает преходящая блокада пресинаптических структур, нарушается холинергическая трансмиссия
связан с воздействием

Слайд 42ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Миастения (Myasthenia gravis)
Заболевание аутоиммунной природы
Вырабатываются антитела к белку ацетилхолиновых рецепторов
ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Миастения (Myasthenia gravis)
Заболевание аутоиммунной природы
Вырабатываются антитела к белку ацетилхолиновых рецепторов

Слайд 43КЛИНИКА МИАСТЕНИИ
Начало, как правило, с глазных мышц (птоз, двоение, сонное выражение лица)
Часто
КЛИНИКА МИАСТЕНИИ
Начало, как правило, с глазных мышц (птоз, двоение, сонное выражение лица)
Часто

Слайд 44КЛИНИЧЕСКИЕ ПРОБЫ, ВЫЯВЛЯЮЩИЕ ПАТОЛОГИЧЕСКУЮ МЫШЕЧНУЮ УТОМЛЯЕМОСТЬ
Фиксировать взор вверх – 30 секунд (больной
КЛИНИЧЕСКИЕ ПРОБЫ, ВЫЯВЛЯЮЩИЕ ПАТОЛОГИЧЕСКУЮ МЫШЕЧНУЮ УТОМЛЯЕМОСТЬ
Фиксировать взор вверх – 30 секунд (больной

Слайд 45ТЕЧЕНИЕ МИАСТЕНИИ
Характерно течение с ремиссиями
В 20% наблюдаются миастенические кризы (генерализованная мышечная слабость,
ТЕЧЕНИЕ МИАСТЕНИИ
Характерно течение с ремиссиями
В 20% наблюдаются миастенические кризы (генерализованная мышечная слабость,

Слайд 46ЛЕЧЕНИЕ МИАСТЕНИИ
Антихолинэстеразные
прозерин – быстрого действия
калимин – медленного
Преднизолон (больным старше 50 лет)
Плазмаферез
Тимэктомия
При
ЛЕЧЕНИЕ МИАСТЕНИИ
Антихолинэстеразные
прозерин – быстрого действия
калимин – медленного
Преднизолон (больным старше 50 лет)
Плазмаферез
Тимэктомия
При

Слайд 47МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Синдром Ламберта-Итона –
паранеопластический синдром
Наблюдается у мужчин старше 40 лет при
МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Синдром Ламберта-Итона –
паранеопластический синдром
Наблюдается у мужчин старше 40 лет при

Слайд 48МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Клиника
не страдают глазные мышцы
нет реакции на антихолинэстеразные препараты
миастенический синдром может опережать
МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Клиника
не страдают глазные мышцы
нет реакции на антихолинэстеразные препараты
миастенический синдром может опережать

Слайд 49МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Другие причины
Заболевания щитовидной железы (аутоиммунные)
Интоксикация лекарственными препаратами:
неомицин
гентамицин
Д-пенициламин (при
МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Другие причины
Заболевания щитовидной железы (аутоиммунные)
Интоксикация лекарственными препаратами:
неомицин
гентамицин
Д-пенициламин (при

Слайд 50БОЛЕЗНИ, СВЯЗАННЫЕ С ПОРАЖЕНИЕМ МЫШЦ (МИОПАТИИ)
Общие признаки:
Отсутствие чувствительных и вегетативных нарушений
Поражение проксимальных
БОЛЕЗНИ, СВЯЗАННЫЕ С ПОРАЖЕНИЕМ МЫШЦ (МИОПАТИИ)
Общие признаки:
Отсутствие чувствительных и вегетативных нарушений
Поражение проксимальных

Слайд 51ПОЛИМИОЗИТ
Не наследственное (аутоиммунное) заболевание
Нарушение клеточного и гуморального иммунитета («воспалительная миопатия»)
Может протекать с
ПОЛИМИОЗИТ
Не наследственное (аутоиммунное) заболевание
Нарушение клеточного и гуморального иммунитета («воспалительная миопатия»)
Может протекать с

Слайд 52КЛИНИКА ПОЛИМИОЗИТА
Начало – острое или подострое
Мышечная слабость
Недомогание, артралгии, миалгии
Повышение температуры тела
Поражение проксимальных
КЛИНИКА ПОЛИМИОЗИТА
Начало – острое или подострое
Мышечная слабость
Недомогание, артралгии, миалгии
Повышение температуры тела
Поражение проксимальных

Слайд 53ПОЛИМИОЗИТ
Данные дополнительного исследования:
Уровень КФК не коррелирует с тяжестью
↑ миоглобин в сыворотке крови
↑СОЭ
Биопсия
ПОЛИМИОЗИТ
Данные дополнительного исследования:
Уровень КФК не коррелирует с тяжестью
↑ миоглобин в сыворотке крови
↑СОЭ
Биопсия

Слайд 54
ВОСПАЛИТЕЛЬНАЯ МИОПАТИЯ (ПОЛИМИОЗИТ)
I
Биопсия мышцы
Воспалительные изменения в мышце
ВОСПАЛИТЕЛЬНАЯ МИОПАТИЯ (ПОЛИМИОЗИТ)
I
Биопсия мышцы
Воспалительные изменения в мышце

Слайд 55НАСЛЕДСТВЕННЫЕ МИОДИСТРОФИИ (МИОПАТИИ)
Известно много форм и вариантов
Псевдогипертрофическая миодистрофия
Дюшена
Миодистрофия Ландузи-Дежерина
НАСЛЕДСТВЕННЫЕ МИОДИСТРОФИИ (МИОПАТИИ)
Известно много форм и вариантов
Псевдогипертрофическая миодистрофия
Дюшена
Миодистрофия Ландузи-Дежерина

Слайд 56
Неравномерность диаметра мышечных волокон, разрастание соединительной и жировой тканей
ПЕРВИЧНЫЕ АМИОТРОФИИ
Неравномерность диаметра мышечных волокон, разрастание соединительной и жировой тканей
ПЕРВИЧНЫЕ АМИОТРОФИИ

Слайд 57ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МИОДИСТРОФИЯ ДЮШЕНА
Описана в 1861 году Дюшеном
В 1879 году Говерс обобщил материал:
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МИОДИСТРОФИЯ ДЮШЕНА
Описана в 1861 году Дюшеном
В 1879 году Говерс обобщил материал:

Слайд 58КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
Первые признаки появляются с момента начала ходьбы (близко колени,
КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
Первые признаки появляются с момента начала ходьбы (близко колени,

Слайд 59КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
Умственная отсталость – 30%.
ЭМГ – признаки первично-мышечного поражения.
Биопсия мышц
КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
Умственная отсталость – 30%.
ЭМГ – признаки первично-мышечного поражения.
Биопсия мышц

Слайд 60КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
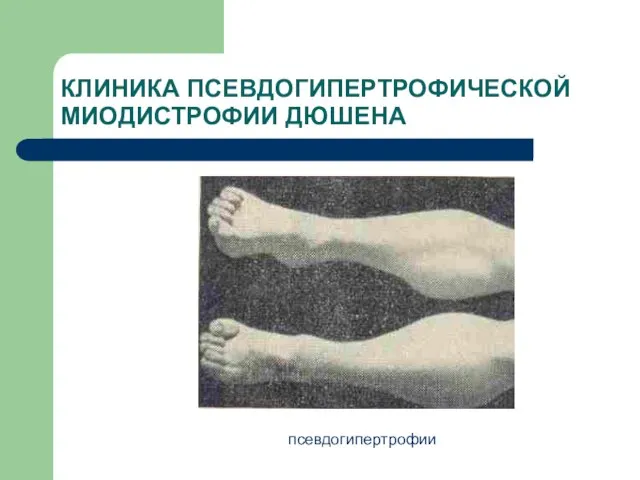
псевдогипертрофии
КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
псевдогипертрофии
Слайд 61
Мальчик 5 лет
Наблюдаются псевдогипертрофии мышц, лордоз
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ ФОРМА ДЮШЕНА
Мальчик 5 лет
Наблюдаются псевдогипертрофии мышц, лордоз
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ ФОРМА ДЮШЕНА

Слайд 62СИМПТОМ «ВСТАВАНИЯ ЛЕСЕНКОЙ» ПРИ МИОПАТИИ
СИМПТОМ «ВСТАВАНИЯ ЛЕСЕНКОЙ» ПРИ МИОПАТИИ

Слайд 63МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Описана в 1884 -1886 г.г. (Дежерин, Ландузи)
Частота встречаемости–0,4 на 100 000
МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Описана в 1884 -1886 г.г. (Дежерин, Ландузи)
Частота встречаемости–0,4 на 100 000

Слайд 64КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Дебют в 15-25 лет (≈20 лет)
Слабость и атрофии мышц лица,
КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Дебют в 15-25 лет (≈20 лет)
Слабость и атрофии мышц лица,

Слайд 65КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Слабость мышц лица:
глазные щели не смыкаются ночью
больные не могут
КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Слабость мышц лица:
глазные щели не смыкаются ночью
больные не могут

Слайд 66КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Атрофия и слабость мышц плечевого и тазового поясов:
крыловидные лопатки, гипотрофия
КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Атрофия и слабость мышц плечевого и тазового поясов:
крыловидные лопатки, гипотрофия

Слайд 67КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Интеллект не страдает
КФК↑ у 50 - 80%
ЛДГ↑ у 20%
Альдолаза↑ у
КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Интеллект не страдает
КФК↑ у 50 - 80%
ЛДГ↑ у 20%
Альдолаза↑ у

Слайд 68
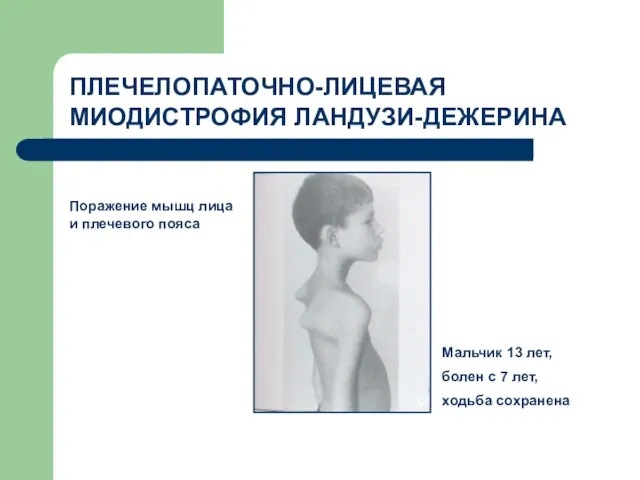
ПЛЕЧЕЛОПАТОЧНО-ЛИЦЕВАЯ МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Поражение мышц лица и плечевого пояса
Мальчик 13 лет,
болен
ПЛЕЧЕЛОПАТОЧНО-ЛИЦЕВАЯ МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Поражение мышц лица и плечевого пояса
Мальчик 13 лет,
болен
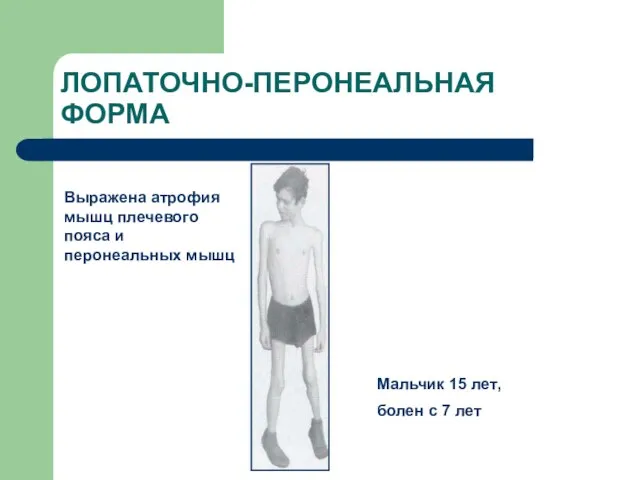
Слайд 69ЛОПАТОЧНО-ПЕРОНЕАЛЬНАЯ ФОРМА
Выражена атрофия мышц плечевого пояса и перонеальных мышц
Мальчик 15 лет,
ЛОПАТОЧНО-ПЕРОНЕАЛЬНАЯ ФОРМА
Выражена атрофия мышц плечевого пояса и перонеальных мышц
Мальчик 15 лет,
Слайд 70МИОДИСТРОФИЯ ЭРБА-РОТА
Описана в 1884 году Эрбом
Тип наследования аутосомно-рецессивный
Экспрессивность гена у членов семьи
МИОДИСТРОФИЯ ЭРБА-РОТА
Описана в 1884 году Эрбом
Тип наследования аутосомно-рецессивный
Экспрессивность гена у членов семьи

Слайд 71КЛИНИКА МИОДИСТРОФИИ ЭРБА
Дебют во 2-м десятилетии, но может быть и в детстве
КЛИНИКА МИОДИСТРОФИИ ЭРБА
Дебют во 2-м десятилетии, но может быть и в детстве

Слайд 72КЛИНИКА МИОДИСТРОФИИ ЭРБА
Форма Лейдена-Мебиуса
начало с проксимальных отделов ног
Форма Эрба
начало с
КЛИНИКА МИОДИСТРОФИИ ЭРБА
Форма Лейдена-Мебиуса
начало с проксимальных отделов ног
Форма Эрба
начало с

Слайд 73
ТАЗОВО-БЕДРЕННАЯ ФОРМА МИОПАТИИ ЭРБА
Больной 17 лет,
болен с 6 лет
Атрофия
ТАЗОВО-БЕДРЕННАЯ ФОРМА МИОПАТИИ ЭРБА
Больной 17 лет,
болен с 6 лет
Атрофия
 Строевая стойка с оружием
Строевая стойка с оружием Правление Петра I
Правление Петра I Отечественная_война_1812_года
Отечественная_война_1812_года Региональный бюджет, финансовая и налоговая политика
Региональный бюджет, финансовая и налоговая политика HR Мотивация персонала
HR Мотивация персонала Подпотолочная роспись, Государственный Эрмитаж, Зал 121. К письменной экзаменационной работе
Подпотолочная роспись, Государственный Эрмитаж, Зал 121. К письменной экзаменационной работе Социальная работа с разными группами населения
Социальная работа с разными группами населения Оборотные средства организации
Оборотные средства организации Машины
Машины МОУ-Клинская средняя школа № 17Эссе по обществознанию:ТЕОРИЯ И ПРАКТИКААвтор: Романова Валентина Александровна
МОУ-Клинская средняя школа № 17Эссе по обществознанию:ТЕОРИЯ И ПРАКТИКААвтор: Романова Валентина Александровна Prilozhenie_1_k_individualnomu_zadaniyu_po_PM_01_shablon_dlya_zapolneniya
Prilozhenie_1_k_individualnomu_zadaniyu_po_PM_01_shablon_dlya_zapolneniya Сергей Есенин
Сергей Есенин Говорю «СПАСИБО»
Говорю «СПАСИБО» Деньги Сомали
Деньги Сомали Патент
Патент Презентация на тему Действия с векторами
Презентация на тему Действия с векторами Производственная структура предприятия
Производственная структура предприятия Романское искусство и его примеры
Романское искусство и его примеры Арктика
Арктика Гражданские правоотношения
Гражданские правоотношения Фруктовые деревья
Фруктовые деревья Невесомость предсказаная и неожиданная
Невесомость предсказаная и неожиданная Звук. Звуковые явления
Звук. Звуковые явления Логические основы компьютера
Логические основы компьютера Решение уравнений третьей степени
Решение уравнений третьей степени Реализация региональной программы профориентационной работы в Костромской области
Реализация региональной программы профориентационной работы в Костромской области Синтаксис коррелятивных конструкций русского языка с позиции генеративной грамматики
Синтаксис коррелятивных конструкций русского языка с позиции генеративной грамматики Сравнительный анализ стоимости и срока службы трамвайных переездов (на перекрестках)
Сравнительный анализ стоимости и срока службы трамвайных переездов (на перекрестках)