- Термодинамика однокомпонентных систем

Содержание
- 3. Реальные газы. Летучесть Уравнения для термодинамических потенциалов реальных газов можно получить на основании уравнений состояния. Однако
- 4. Широко известно уравнение Ван-дер-Ваальса: (р + а/V2)(V –b) = RT, (11) которое характеризует не только изменения
- 5. Гилберт Льюис предложил метод расчета термодинамических характеристик реальных газов, основанный на том, что для них формально
- 7. Изотермы реальных газов (Т1
- 9. Зависимость объемной поправки от давления (Т1
- 11. Стандартное состояние Изобарный потенциал реального газа выражается уравнением (13), в котором G(T) – постоянная интегрирования, абсолютную
- 12. Переход газа из любого состояния в стандартное схематически изображен на рис. Если реальный газ находится при
- 13. Переход в стандартное состояние (1 – изотерма реального газа, 2 – изотерма идеального газа)
- 17. Фазовые превращения. Уравнение Клаузиуса – Клапейрона. Индивидуальное вещество может находиться в газообразном, жидком или твердом (в
- 23. Более точным является правило Гильдебранда, согласно которому энтропии испарения жидкостей равны между собой при температурах, для
- 24. Диаграммы состояния Индивидуальное вещество может существовать в нескольких агрегатных состояниях, т. е. образовывать несколько фаз. Например,
- 25. В общем случае нам не известны уравнения состояния отдельных фаз, поэтому зависимость между переменными, определяющими состояние
- 26. Совокупность этих точек дает диаграмму (рис.), состоящую из нескольких поверхностей, определенным образом расположенных в пространстве. Точки
- 27. Схематическое изображение объемной диаграммы состояния однокомпонентной системы и ее проекций на плоскости
- 29. Скачать презентацию

Слайд 3Реальные газы. Летучесть
Уравнения для термодинамических потенциалов реальных газов можно получить на основании
Реальные газы. Летучесть
Уравнения для термодинамических потенциалов реальных газов можно получить на основании

Слайд 4Широко известно уравнение Ван-дер-Ваальса:
(р + а/V2)(V –b) = RT, (11)
которое характеризует
Широко известно уравнение Ван-дер-Ваальса:
(р + а/V2)(V –b) = RT, (11)
которое характеризует

Слайд 5Гилберт Льюис предложил метод расчета термодинамических характеристик реальных газов, основанный на том,
Гилберт Льюис предложил метод расчета термодинамических характеристик реальных газов, основанный на том,


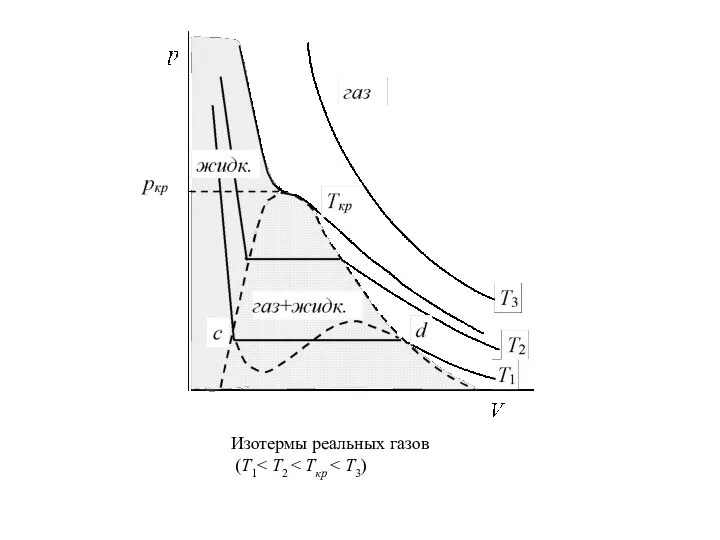
Слайд 7Изотермы реальных газов
(Т1< Т2 < Ткр < Т3)
Изотермы реальных газов
(Т1< Т2 < Ткр < Т3)

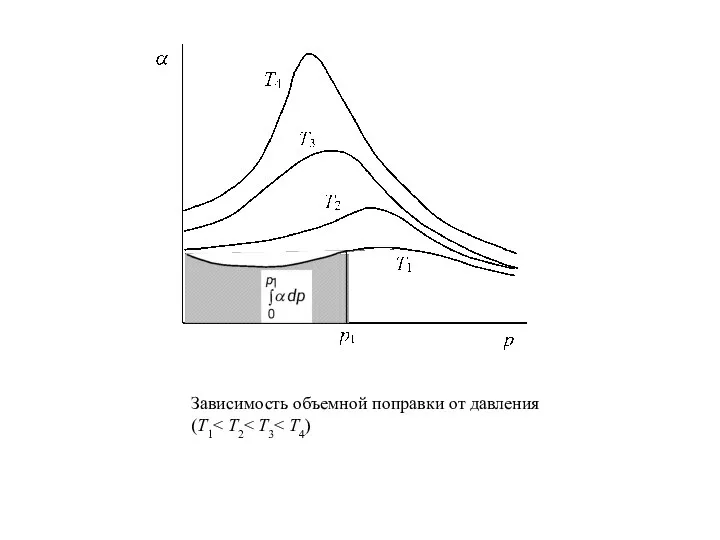
Слайд 9Зависимость объемной поправки от давления (Т1< Т2< Т3< Т4)
Зависимость объемной поправки от давления (Т1< Т2< Т3< Т4)

Слайд 11Стандартное состояние
Изобарный потенциал реального газа выражается уравнением (13), в котором G(T) –
Стандартное состояние
Изобарный потенциал реального газа выражается уравнением (13), в котором G(T) –

Слайд 12Переход газа из любого состояния в стандартное схематически изображен на рис.
Если
Переход газа из любого состояния в стандартное схематически изображен на рис.
Если

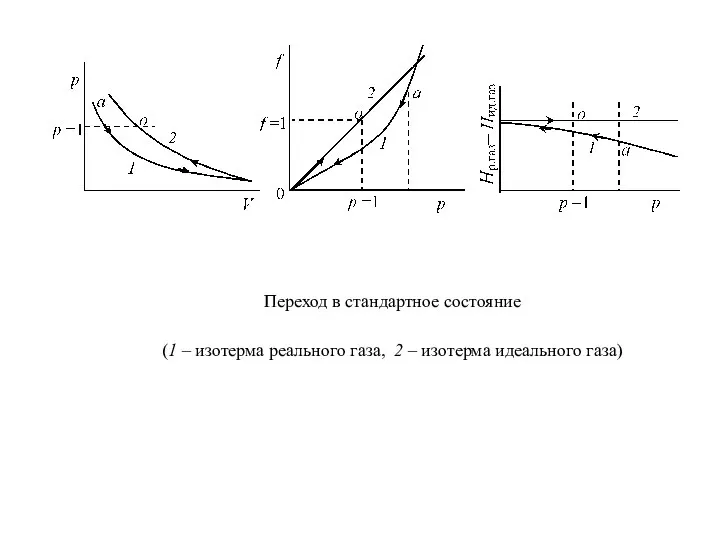
Слайд 13Переход в стандартное состояние
(1 – изотерма реального газа, 2 – изотерма
Переход в стандартное состояние
(1 – изотерма реального газа, 2 – изотерма



Слайд 17Фазовые превращения. Уравнение Клаузиуса – Клапейрона.
Индивидуальное вещество может находиться в газообразном, жидком
Фазовые превращения. Уравнение Клаузиуса – Клапейрона.
Индивидуальное вещество может находиться в газообразном, жидком






Слайд 23Более точным является правило Гильдебранда, согласно которому энтропии испарения жидкостей равны между
Более точным является правило Гильдебранда, согласно которому энтропии испарения жидкостей равны между

Слайд 24Диаграммы состояния
Индивидуальное вещество может существовать в нескольких агрегатных состояниях, т. е. образовывать
Диаграммы состояния
Индивидуальное вещество может существовать в нескольких агрегатных состояниях, т. е. образовывать

Слайд 25В общем случае нам не известны уравнения состояния отдельных фаз, поэтому зависимость
В общем случае нам не известны уравнения состояния отдельных фаз, поэтому зависимость

Слайд 26Совокупность этих точек дает диаграмму (рис.), состоящую из нескольких поверхностей, определенным образом
Совокупность этих точек дает диаграмму (рис.), состоящую из нескольких поверхностей, определенным образом

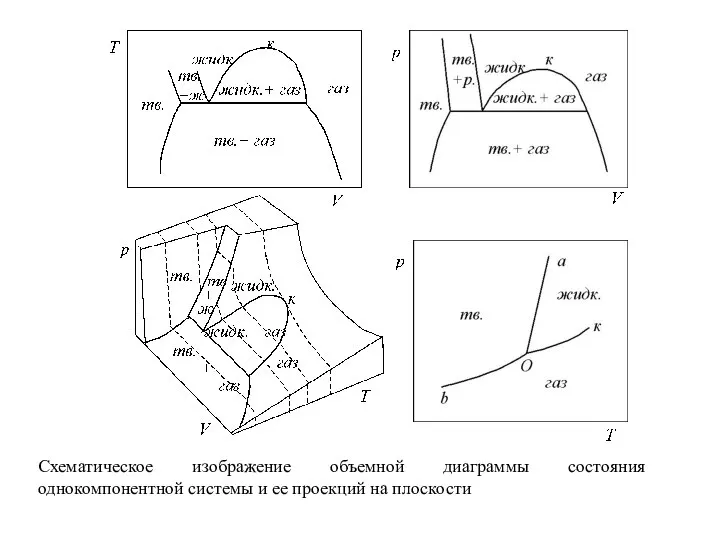
Слайд 27Схематическое изображение объемной диаграммы состояния
однокомпонентной системы и ее проекций на плоскости
Схематическое изображение объемной диаграммы состояния
однокомпонентной системы и ее проекций на плоскости
 Двойное лучепреломление. (Лекция 39)
Двойное лучепреломление. (Лекция 39) Управление вертолетом. Основные моменты
Управление вертолетом. Основные моменты Основные понятия и задачи кинематики (продолжение)
Основные понятия и задачи кинематики (продолжение) Решение задач на равновесие сходящейся системы сил
Решение задач на равновесие сходящейся системы сил Принцип Гюйгенса-Френеля
Принцип Гюйгенса-Френеля Тезаурус по атомной энергетике
Тезаурус по атомной энергетике Система электрического пуска
Система электрического пуска Кипение. Температура кипения
Кипение. Температура кипения Решение задач по теме Термодинамика. Теплообмен. Первое начало термодинамики. Тепловые двигатели
Решение задач по теме Термодинамика. Теплообмен. Первое начало термодинамики. Тепловые двигатели Качество деталей машин. Качество материалов
Качество деталей машин. Качество материалов Презентация на тему Применение закона рычага к блоку
Презентация на тему Применение закона рычага к блоку  Метод наложения для расчета электрических цепей
Метод наложения для расчета электрических цепей Жидкие кристаллы
Жидкие кристаллы ФОМНЭ_2022_Лекция № 2
ФОМНЭ_2022_Лекция № 2 Звуковые колебания. Источники звука (9 класс)
Звуковые колебания. Источники звука (9 класс) Исследование зависимости между массой тела и силой, с которой это тело притягивается Землей
Исследование зависимости между массой тела и силой, с которой это тело притягивается Землей phpvTLe67_sceplenie-traktora-NTZ-80
phpvTLe67_sceplenie-traktora-NTZ-80 Устройство карданной передачи, разработка технологической карты
Устройство карданной передачи, разработка технологической карты Линейчатые оптические спектры. Поглощение и испускание света атомами
Линейчатые оптические спектры. Поглощение и испускание света атомами Устройство и ремонт унифицированной гидропередачи УГП-1200
Устройство и ремонт унифицированной гидропередачи УГП-1200 Тонкостенные сферические сосуды и трубы
Тонкостенные сферические сосуды и трубы Лекция15
Лекция15 Линейка - как измерительный прибор в физике
Линейка - как измерительный прибор в физике Расчет пролетного строения. Расчет плиты балластного корыта. Расчет главной балки.Лекция №7
Расчет пролетного строения. Расчет плиты балластного корыта. Расчет главной балки.Лекция №7 Основы динамики
Основы динамики Расчет стоимости электрической энергии
Расчет стоимости электрической энергии Задачи атомной и ядерной физики, астрономии
Задачи атомной и ядерной физики, астрономии Интеллектуальная игра Что? Где? Когда?
Интеллектуальная игра Что? Где? Когда?