- Determination of optical properties of pentacoordinated silicon complexes using DFT method

Содержание
- 2. Relevance Aims and objectives of the study Aim - To define the optical properties of pentacoordinated
- 3. Theoretical significance Practical significance The research uses a method DFT that allows to get more reliable
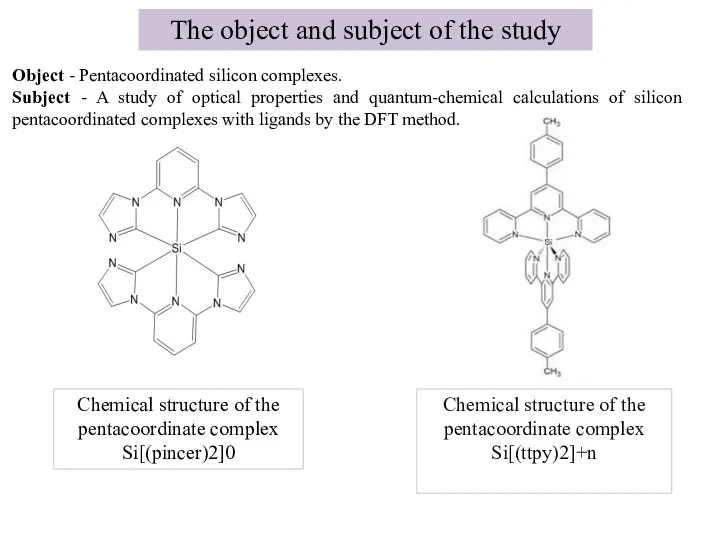
- 4. The object and subject of the study Chemical structure of the pentacoordinate complex Si[(ttpy)2]+n Chemical structure
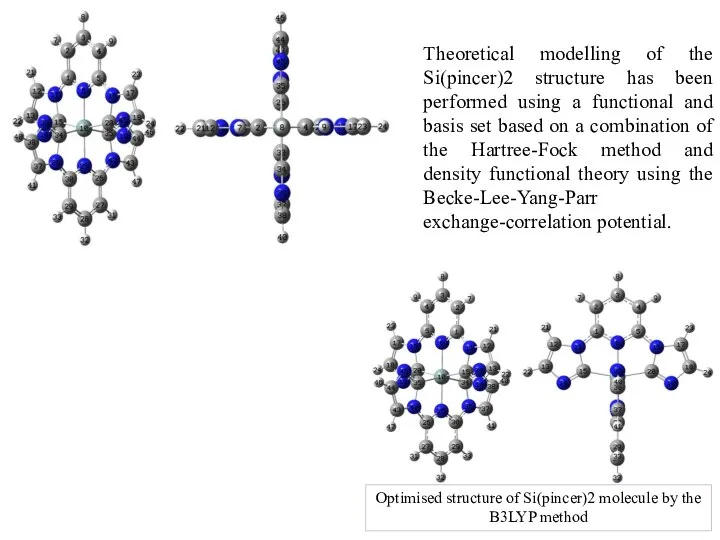
- 6. Optimised structure of Si(pincer)2 molecule by the B3LYP method Theoretical modelling of the Si(pincer)2 structure has
- 7. Electron absorption spectrum graph for [Si(pincer)2]0 structure TD-DFT calculations reproduce the observed electron spectrum
- 8. Molecular orbitals involved in intensive transitions of the [Si(pincer)2]0 structure HOMO - Highest occupied MO LUMO
- 9. Finite fully optimised structures Si[(ttpy)2]+n
- 10. Electronic absorption spectra of the complex [Si(ttpy)2]+n : blue line, n = 4 ; red line,
- 11. Molecular orbitals involved in intensive transitions of the [Si(ttpy)2]+4 structure
- 12. Molecular orbitals involved in intense transitions of the [Si(ttpy)2]+2 structure
- 13. Molecular orbitals involved in intensive transitions of the [Si(ttpy)2]0 structure
- 14. The spectra of the [Si(ttpy)2]+n states are electrochemically generated at -0.245 V (blue line, n =
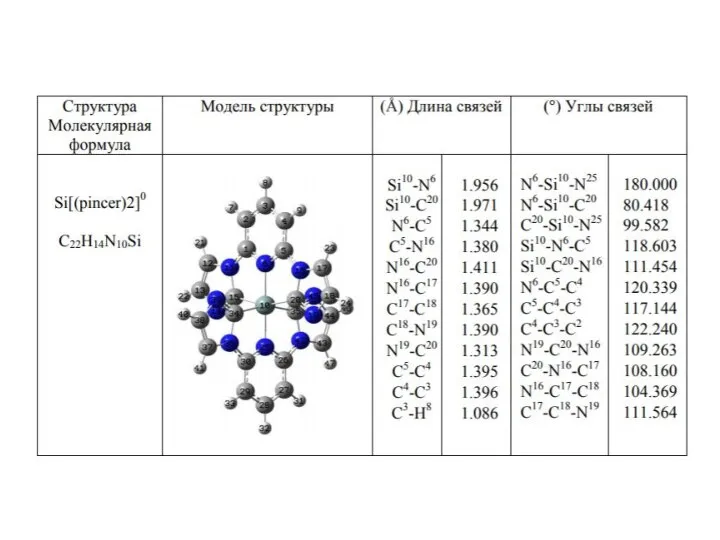
- 15. Quantum-chemical calculation of the structural parameters of [Si(pincer)2]0 ; [Si(ttpy)2]+4 ; [Si(ttpy)2] +2 ; [Si(ttpy)2] 0
- 17. Скачать презентацию

Слайд 3Theoretical significance
Practical significance
The research uses a method DFT that allows to get
Theoretical significance
Practical significance
The research uses a method DFT that allows to get

Слайд 4The object and subject of the study
Chemical structure of the pentacoordinate complex
The object and subject of the study
Chemical structure of the pentacoordinate complex
Слайд 6Optimised structure of Si(pincer)2 molecule by the B3LYP method
Theoretical modelling of the
Optimised structure of Si(pincer)2 molecule by the B3LYP method
Theoretical modelling of the
Слайд 7Electron absorption spectrum graph for [Si(pincer)2]0 structure
TD-DFT calculations reproduce the observed electron
Electron absorption spectrum graph for [Si(pincer)2]0 structure
TD-DFT calculations reproduce the observed electron
![Electron absorption spectrum graph for [Si(pincer)2]0 structure TD-DFT calculations reproduce the observed electron spectrum](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-6.jpg)
Слайд 8Molecular orbitals involved in intensive transitions of the [Si(pincer)2]0 structure
HOMO - Highest
Molecular orbitals involved in intensive transitions of the [Si(pincer)2]0 structure
HOMO - Highest
![Molecular orbitals involved in intensive transitions of the [Si(pincer)2]0 structure HOMO -](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-7.jpg)
Слайд 9Finite fully optimised structures Si[(ttpy)2]+n
Finite fully optimised structures Si[(ttpy)2]+n
![Finite fully optimised structures Si[(ttpy)2]+n](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-8.jpg)
Слайд 10Electronic absorption spectra of the complex [Si(ttpy)2]+n :
blue line, n =
Electronic absorption spectra of the complex [Si(ttpy)2]+n :
blue line, n =
![Electronic absorption spectra of the complex [Si(ttpy)2]+n : blue line, n =](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-9.jpg)
Слайд 11Molecular orbitals involved in intensive transitions of the [Si(ttpy)2]+4 structure
Molecular orbitals involved in intensive transitions of the [Si(ttpy)2]+4 structure
![Molecular orbitals involved in intensive transitions of the [Si(ttpy)2]+4 structure](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-10.jpg)
Слайд 12Molecular orbitals involved in intense transitions of the [Si(ttpy)2]+2 structure
Molecular orbitals involved in intense transitions of the [Si(ttpy)2]+2 structure
![Molecular orbitals involved in intense transitions of the [Si(ttpy)2]+2 structure](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-11.jpg)
Слайд 13Molecular orbitals involved in intensive transitions of the [Si(ttpy)2]0 structure
Molecular orbitals involved in intensive transitions of the [Si(ttpy)2]0 structure
![Molecular orbitals involved in intensive transitions of the [Si(ttpy)2]0 structure](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-12.jpg)
Слайд 14The spectra of the [Si(ttpy)2]+n states are electrochemically generated
at -0.245 V
The spectra of the [Si(ttpy)2]+n states are electrochemically generated
at -0.245 V
![The spectra of the [Si(ttpy)2]+n states are electrochemically generated at -0.245 V](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-13.jpg)
Слайд 15 Quantum-chemical calculation of the structural parameters of [Si(pincer)2]0 ; [Si(ttpy)2]+4 ;
Quantum-chemical calculation of the structural parameters of [Si(pincer)2]0 ; [Si(ttpy)2]+4 ;
![Quantum-chemical calculation of the structural parameters of [Si(pincer)2]0 ; [Si(ttpy)2]+4 ; [Si(ttpy)2]](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1112580/slide-14.jpg)
 Химическая география чудес
Химическая география чудес Эфирные масла
Эфирные масла Получение этиленгликоля
Получение этиленгликоля Периодическая система химических элементов Д.И. Меделеева
Периодическая система химических элементов Д.И. Меделеева Исследование интерактивной компьютерной химической модели
Исследование интерактивной компьютерной химической модели Презентация на тему Неметаллы
Презентация на тему Неметаллы  Изучение свойств пластичных масс для лепки
Изучение свойств пластичных масс для лепки Отжиг сталей (отжиг 2-го рода) Лекция 2
Отжиг сталей (отжиг 2-го рода) Лекция 2 Призер муниципального єтапа всероссийской олимпиады школьников по химии Карасева Светлана
Призер муниципального єтапа всероссийской олимпиады школьников по химии Карасева Светлана Химический состав клетки
Химический состав клетки Алкены. Пентен- С5Н10
Алкены. Пентен- С5Н10 Дисперсные системы
Дисперсные системы Презентация 1
Презентация 1 Вода - растворитель
Вода - растворитель Алюминий. Применение
Алюминий. Применение Окислительно-восстановительные реакции. ОГЭ по химии, задание 15
Окислительно-восстановительные реакции. ОГЭ по химии, задание 15 Ароматические углеводороды - Арены
Ароматические углеводороды - Арены Реакции ионного обмена. Электролитическая диссоциация
Реакции ионного обмена. Электролитическая диссоциация Марганец. Калий
Марганец. Калий Изменения в составе ядра атома Задачи урока: Познакомиться с понятием «ядерные процессы», «изотопы» Развить понятие «Химически
Изменения в составе ядра атома Задачи урока: Познакомиться с понятием «ядерные процессы», «изотопы» Развить понятие «Химически Органические вещества. 9 класс
Органические вещества. 9 класс Арены. Бензол. 9 класс
Арены. Бензол. 9 класс Ковалентная химическая связь
Ковалентная химическая связь Определение количества этилендиамина в соединении [Gd(en)x][Fe(CN)6]
Определение количества этилендиамина в соединении [Gd(en)x][Fe(CN)6] Основи прийняття рішень, щодо захисту населення під час різних фаз радіаційної аварії. Критерії для прийняття рішень
Основи прийняття рішень, щодо захисту населення під час різних фаз радіаційної аварії. Критерії для прийняття рішень Устойчивость коллоидных лиофобных дисперсных систем. Теория ДЛФО. Лекция 14
Устойчивость коллоидных лиофобных дисперсных систем. Теория ДЛФО. Лекция 14 Взаимное притяжение и отталкивание молекул
Взаимное притяжение и отталкивание молекул Электрические явления на поверхности раздела фаз. Лекция 8
Электрические явления на поверхности раздела фаз. Лекция 8