- IMMUNOPATOLOGIYa_pptx1

Содержание
- 2. * Физиологическая форма иммуногенной реактивности. * Формируется в результате реализации наследуемой генетической программы и/или при контакте
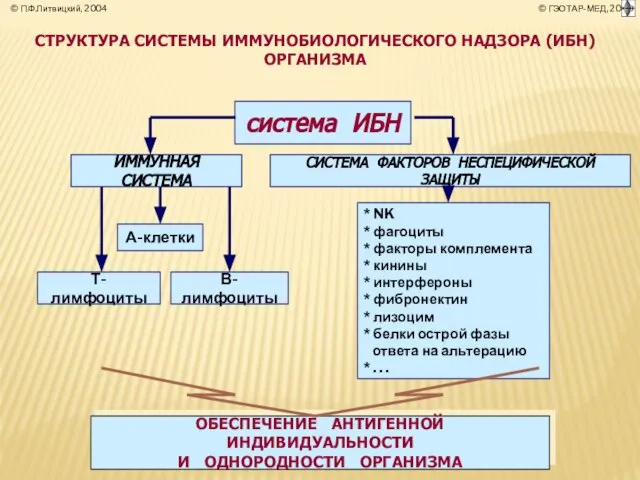
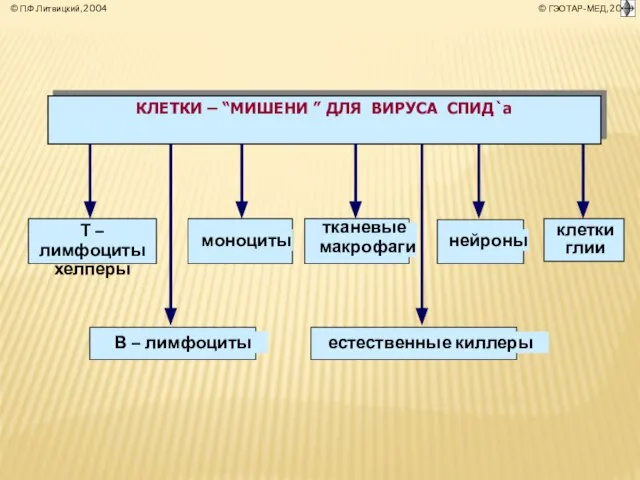
- 4. СТРУКТУРА СИСТЕМЫ ИММУНОБИОЛОГИЧЕСКОГО НАДЗОРА (ИБН) ОРГАНИЗМА система ИБН ОБЕСПЕЧЕНИЕ АНТИГЕННОЙ ИНДИВИДУАЛЬНОСТИ И ОДНОРОДНОСТИ ОРГАНИЗМА
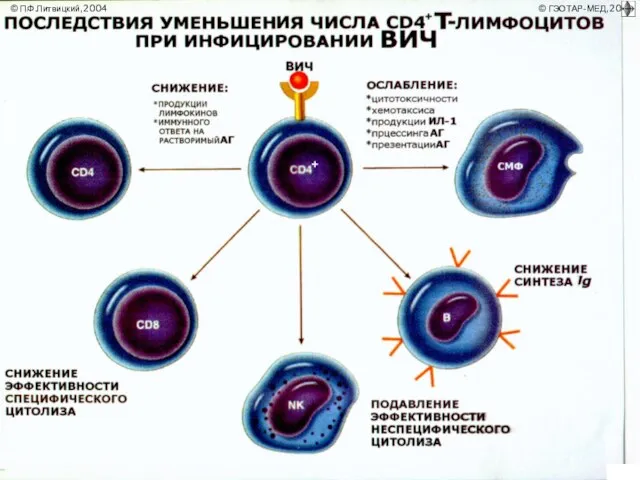
- 6. ИММУНОДЕФИЦИТЫ Иммунодефицит, или иммунологическая недостаточность, — состояние, развивающееся при нарушении иммунных механизмов
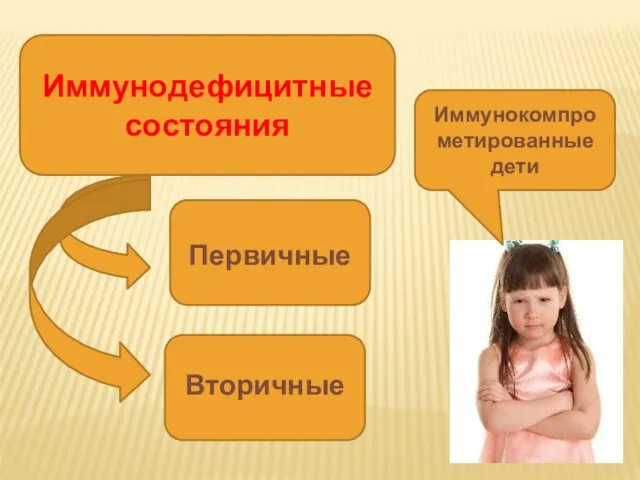
- 7. Иммунодефицитные состояния Первичные Вторичные Иммунокомпрометированные дети
- 8. хроническое или рецидивирующее течение, склонность к прогрессированию политопность (множественные поражения различных органов и тканей) полиэтиологичность (восприимчивость
- 9. Первичные ИДС являются довольно редкими заболеваниями, частота их встречаемости соответствует 1 случаю на 23 000-100 000
- 10. тяжелое, особенно рецидивирующее гнойное заболевание; парапроктит, аноректальный свищ; наличие упорного кандидоза полости рта (молочницы) или других
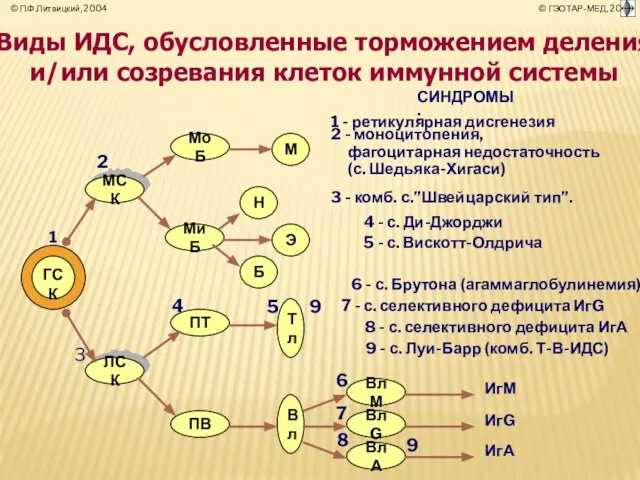
- 11. РЕТИКУЛЯРНАЯ ДИСГЕНЕЗИЯ Ретикулярная дисгенезия — редкое заболевание, характеризующееся дефицитом лимфоцитов и полиморфно-ядерных лейкоцитов. У больных в
- 12. ЧЕДИАКА ХИГАСИ СИНДРОМ Впервые в клинической практики данный синдром был выделен в 1943 году, однако генетический
- 13. СИНДРОМ ЧЕДИАКА-ХИГАСИ
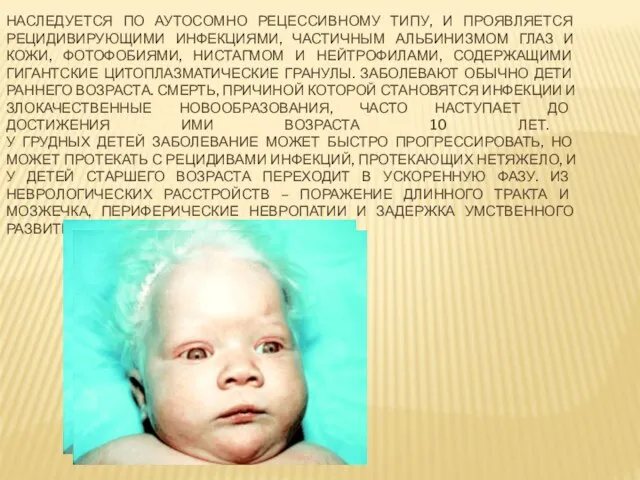
- 14. НАСЛЕДУЕТСЯ ПО АУТОСОМНО РЕЦЕССИВНОМУ ТИПУ, И ПРОЯВЛЯЕТСЯ РЕЦИДИВИРУЮЩИМИ ИНФЕКЦИЯМИ, ЧАСТИЧНЫМ АЛЬБИНИЗМОМ ГЛАЗ И КОЖИ, ФОТОФОБИЯМИ, НИСТАГМОМ
- 15. Нарушение пролиферации, дифференцировки, хемотаксиса нейтрофилов и макрофагов и самого процесса фагоцитоза Синдром гипериммуноглобулинемии Е, болезнь Костманна,
- 16. ШВЕЙЦАРСКИЙ ТИП ИДС Тяжелый комбинированный иммунодефицит, характеризующийся дефектом клеточного и гуморального иммунитета. Впервые описан Е. Glanzmann
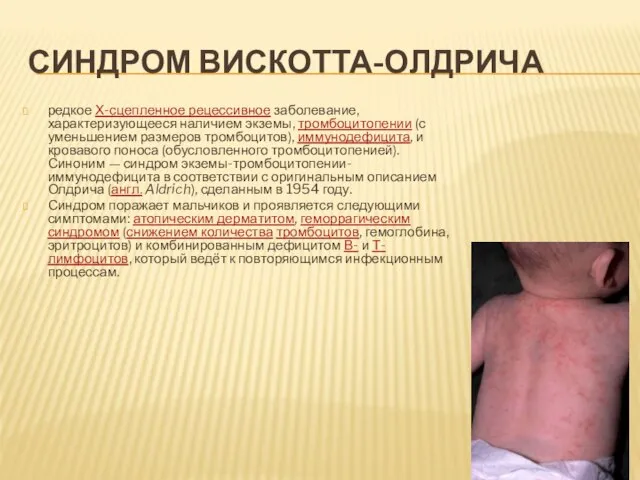
- 17. СИНДРОМ ВИСКОТТА-ОЛДРИЧА редкое Х-сцепленное рецессивное заболевание, характеризующееся наличием экземы, тромбоцитопении (с уменьшением размеров тромбоцитов), иммунодефицита, и
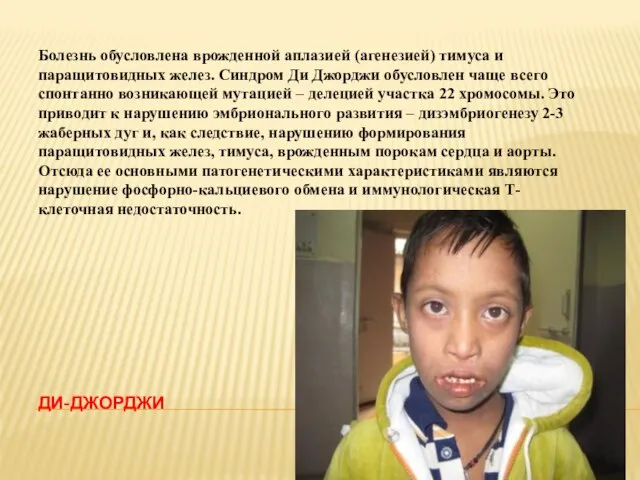
- 18. ДИ-ДЖОРДЖИ Болезнь обусловлена врожденной аплазией (агенезией) тимуса и паращитовидных желез. Синдром Ди Джорджи обусловлен чаще всего
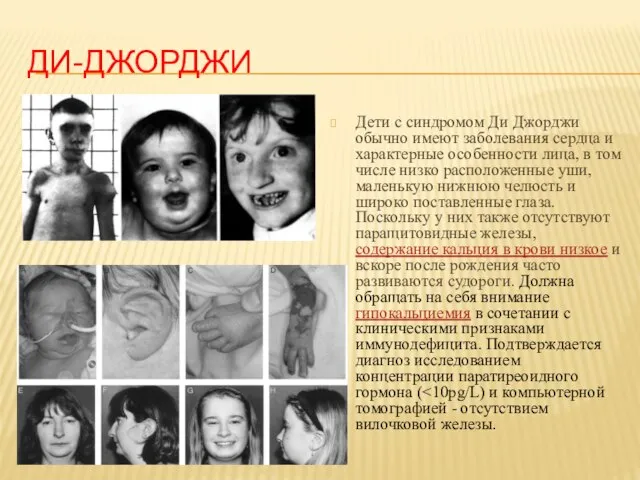
- 19. ДИ-ДЖОРДЖИ Дети с синдромом Ди Джорджи обычно имеют заболевания сердца и характерные особенности лица, в том
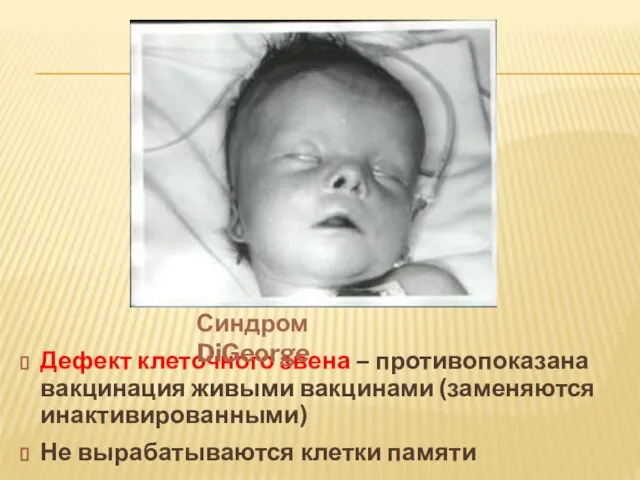
- 20. Дефект клеточного звена – противопоказана вакцинация живыми вакцинами (заменяются инактивированными) Не вырабатываются клетки памяти Синдром DiGeorge
- 21. СИНДРОМ БРУТОНА МУТАНТНЫЙ БЕЛОК — ТИРОЗИНКИНАЗА БРУТОНА. МУТАНТНЫЙ ГЕН ВТК КАРТИРОВАН НА XQ21.3—22.2. Впервые случай заболевания
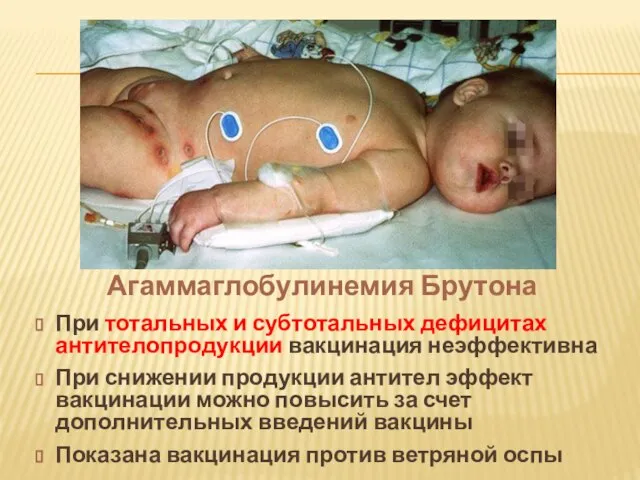
- 22. При тотальных и субтотальных дефицитах антителопродукции вакцинация неэффективна При снижении продукции антител эффект вакцинации можно повысить
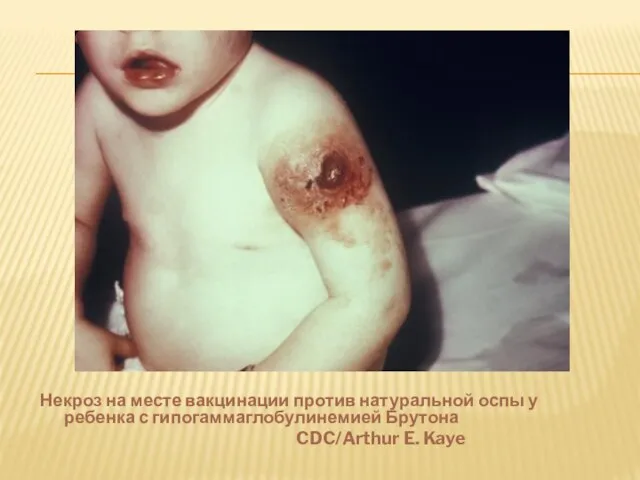
- 23. Некроз на месте вакцинации против натуральной оспы у ребенка с гипогаммаглобулинемией Брутона CDC/Arthur E. Kaye
- 24. БРУТОНА СИНДРОМ Первые симптомы заболевания проявляются, как правило, в возрасте менее 1 года, чаще всего после
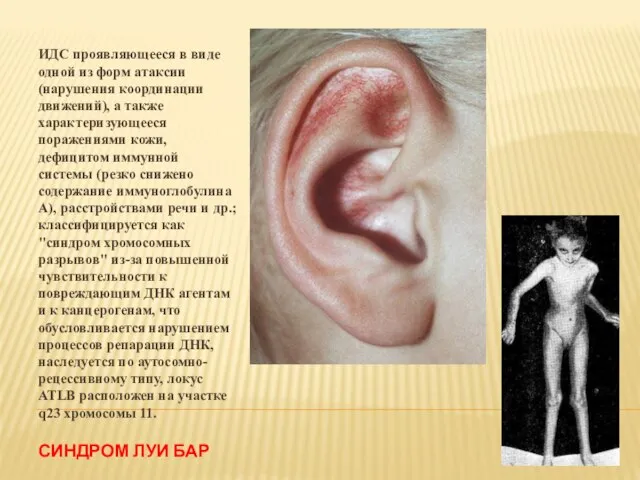
- 25. СИНДРОМ ЛУИ БАР ИДС проявляющееся в виде одной из форм атаксии (нарушения координации движений), а также
- 26. ЛУИ-БАР СИНДРОМ Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у 1/3 пациентов наблюдается
- 27. ИОВА СИНДРОМ (по имени библейского персонажа, пораженного «проказою лютою от подошвы ноги его по самое темя
- 28. ОБЩИЕ ПРИНЦИПЫ При тотальных и субтотальных дефицитах антителопродукции вакцинация неэффективна Дефект клеточного звена – противопоказана вакцинация
- 29. ПРИОБРЕТЕННЫЕ ИДС Обусловлены качественным и количественным голоданием (недостатком белков, витаминов, микроэлементов, Fe, Zn, Cu и др.),
- 30. ПЕРЕЧЕНЬ ОСНОВНЫХ ЗАБОЛЕВАНИЙ, СОПРОВОЖДАЮЩИХСЯ ВТОРИЧНЫМ ИММУНОДЕФИЦИТОМ (ВОЗ) Инфекционные заболевания: Протозойные и глистные болезни - малярия, токсоплазмоз,
- 31. Нарушения питания: истощение, кахексия, нарушения кишечного всасывания и др. Экзогенные и эндогенные интоксикации - при почечной
- 32. Действие различных видов излучения, особенно ионизирующей радиации. Сильные, длительные стрессорные воздействия. Действие лекарственных препаратов (иммунодепрессанты, кортикостероиды,
- 33. КЛИНИЧЕСКИЕ ПРИЗНАКИ ИММУНОДЕФИЦИТНОГО СОСТОЯНИЯ: Частые обострения хронических воспалительных заболеваний разной этиологии Частые обострения герпетической инфекции Длительный
- 36. САРКОМА КАПОШИ
- 37. ГЛАВНЫЙ КОМПЛЕКС ГИСТОСОВМЕСТИМОСТИ – ЭТО ГРУППА ГЕНОВ И КОДИРУЕМЫХ ИМИ АНТИГЕНОВ КЛЕТОЧНОЙ ПОВЕРХНОСТИ, КОТОРЫЕ ИГРАЮТ ВАЖНЕЙШУЮ
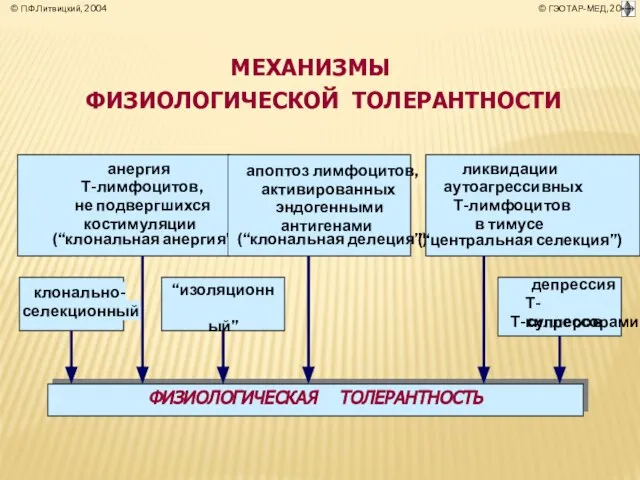
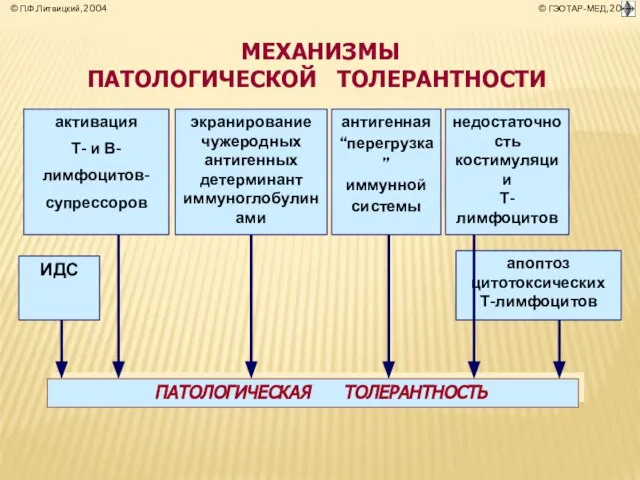
- 38. ПАТОЛОГИЧЕСКАЯ Т О Л Е Р А Н Т Н О С Т Ь (лат. tolerantia
- 43. РТПХ РАНТ-болезнь в эксперименте.
- 46. ФОРМИРОВАНИЕ ТОЛЕРАНТНОСТИ К ТРАНСПЛАНТАТУ Неспецифические методы: 1. Подавление иммунологической реактивности организма реципиента с помощью иммунодепрессантов (циклоспоринА,
- 47. ПАТОГЕНЕЗ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ 1) нарушение физиологической изоляции органов и тканей, по отношению к которым иммунологическая толерантность
- 48. МИАСТЕНИЯ ГРАВИС
- 49. СИНДРОМ ДРЕССЛЕРА Постинфарктное осложнение
- 51. Скачать презентацию
Слайд 2* Физиологическая форма иммуногенной реактивности.
* Формируется в результате реализации наследуемой генетической
программы
* Физиологическая форма иммуногенной реактивности.
* Формируется в результате реализации наследуемой генетической
программы


Слайд 4СТРУКТУРА СИСТЕМЫ ИММУНОБИОЛОГИЧЕСКОГО НАДЗОРА (ИБН) ОРГАНИЗМА
система ИБН
ОБЕСПЕЧЕНИЕ АНТИГЕННОЙ ИНДИВИДУАЛЬНОСТИ
И ОДНОРОДНОСТИ
СТРУКТУРА СИСТЕМЫ ИММУНОБИОЛОГИЧЕСКОГО НАДЗОРА (ИБН) ОРГАНИЗМА
система ИБН
ОБЕСПЕЧЕНИЕ АНТИГЕННОЙ ИНДИВИДУАЛЬНОСТИ
И ОДНОРОДНОСТИ
Слайд 6ИММУНОДЕФИЦИТЫ
Иммунодефицит, или иммунологическая недостаточность, — состояние, развивающееся при нарушении иммунных механизмов
ИММУНОДЕФИЦИТЫ
Иммунодефицит, или иммунологическая недостаточность, — состояние, развивающееся при нарушении иммунных механизмов

Слайд 7Иммунодефицитные состояния
Первичные
Вторичные
Иммунокомпрометированные
дети
Иммунодефицитные состояния
Первичные
Вторичные
Иммунокомпрометированные
дети
Слайд 8хроническое или рецидивирующее течение, склонность к прогрессированию
политопность (множественные поражения различных органов и
хроническое или рецидивирующее течение, склонность к прогрессированию
политопность (множественные поражения различных органов и

Слайд 9 Первичные ИДС являются довольно редкими заболеваниями, частота их встречаемости соответствует 1
Первичные ИДС являются довольно редкими заболеваниями, частота их встречаемости соответствует 1

Слайд 10тяжелое, особенно рецидивирующее гнойное заболевание;
парапроктит, аноректальный свищ;
наличие упорного кандидоза полости рта (молочницы)
тяжелое, особенно рецидивирующее гнойное заболевание;
парапроктит, аноректальный свищ;
наличие упорного кандидоза полости рта (молочницы)

Слайд 11РЕТИКУЛЯРНАЯ ДИСГЕНЕЗИЯ
Ретикулярная дисгенезия — редкое заболевание, характеризующееся дефицитом лимфоцитов и полиморфно-ядерных лейкоцитов.
РЕТИКУЛЯРНАЯ ДИСГЕНЕЗИЯ
Ретикулярная дисгенезия — редкое заболевание, характеризующееся дефицитом лимфоцитов и полиморфно-ядерных лейкоцитов.

Слайд 12ЧЕДИАКА ХИГАСИ СИНДРОМ
Впервые в клинической практики данный синдром был выделен в 1943
ЧЕДИАКА ХИГАСИ СИНДРОМ
Впервые в клинической практики данный синдром был выделен в 1943
Слайд 13СИНДРОМ ЧЕДИАКА-ХИГАСИ
СИНДРОМ ЧЕДИАКА-ХИГАСИ

Слайд 14НАСЛЕДУЕТСЯ ПО АУТОСОМНО РЕЦЕССИВНОМУ ТИПУ, И ПРОЯВЛЯЕТСЯ РЕЦИДИВИРУЮЩИМИ ИНФЕКЦИЯМИ, ЧАСТИЧНЫМ АЛЬБИНИЗМОМ ГЛАЗ
НАСЛЕДУЕТСЯ ПО АУТОСОМНО РЕЦЕССИВНОМУ ТИПУ, И ПРОЯВЛЯЕТСЯ РЕЦИДИВИРУЮЩИМИ ИНФЕКЦИЯМИ, ЧАСТИЧНЫМ АЛЬБИНИЗМОМ ГЛАЗ
Слайд 15Нарушение пролиферации, дифференцировки, хемотаксиса нейтрофилов и макрофагов и самого процесса фагоцитоза
Синдром гипериммуноглобулинемии
Нарушение пролиферации, дифференцировки, хемотаксиса нейтрофилов и макрофагов и самого процесса фагоцитоза
Синдром гипериммуноглобулинемии

Слайд 16ШВЕЙЦАРСКИЙ ТИП ИДС
Тяжелый комбинированный иммунодефицит, характеризующийся дефектом клеточного и гуморального иммунитета. Впервые
ШВЕЙЦАРСКИЙ ТИП ИДС
Тяжелый комбинированный иммунодефицит, характеризующийся дефектом клеточного и гуморального иммунитета. Впервые

Слайд 17СИНДРОМ ВИСКОТТА-ОЛДРИЧА
редкое Х-сцепленное рецессивное заболевание, характеризующееся наличием экземы, тромбоцитопении (с уменьшением
СИНДРОМ ВИСКОТТА-ОЛДРИЧА
редкое Х-сцепленное рецессивное заболевание, характеризующееся наличием экземы, тромбоцитопении (с уменьшением
Слайд 18ДИ-ДЖОРДЖИ
Болезнь обусловлена врожденной аплазией (агенезией) тимуса и паращитовидных желез. Синдром Ди Джорджи
ДИ-ДЖОРДЖИ
Болезнь обусловлена врожденной аплазией (агенезией) тимуса и паращитовидных желез. Синдром Ди Джорджи
Слайд 19ДИ-ДЖОРДЖИ
Дети с синдромом Ди Джорджи обычно имеют заболевания сердца и характерные особенности
ДИ-ДЖОРДЖИ
Дети с синдромом Ди Джорджи обычно имеют заболевания сердца и характерные особенности
Слайд 20Дефект клеточного звена – противопоказана вакцинация живыми вакцинами (заменяются инактивированными)
Не вырабатываются клетки
Дефект клеточного звена – противопоказана вакцинация живыми вакцинами (заменяются инактивированными)
Не вырабатываются клетки
Слайд 21СИНДРОМ БРУТОНА
МУТАНТНЫЙ БЕЛОК — ТИРОЗИНКИНАЗА БРУТОНА. МУТАНТНЫЙ ГЕН ВТК КАРТИРОВАН НА XQ21.3—22.2.
Впервые
СИНДРОМ БРУТОНА
МУТАНТНЫЙ БЕЛОК — ТИРОЗИНКИНАЗА БРУТОНА. МУТАНТНЫЙ ГЕН ВТК КАРТИРОВАН НА XQ21.3—22.2.
Впервые

Слайд 22При тотальных и субтотальных дефицитах антителопродукции вакцинация неэффективна
При снижении продукции антител эффект
При тотальных и субтотальных дефицитах антителопродукции вакцинация неэффективна
При снижении продукции антител эффект
Слайд 23Некроз на месте вакцинации против натуральной оспы у ребенка с гипогаммаглобулинемией Брутона
Некроз на месте вакцинации против натуральной оспы у ребенка с гипогаммаглобулинемией Брутона
Слайд 24БРУТОНА СИНДРОМ
Первые симптомы заболевания проявляются, как правило, в возрасте менее 1 года,
БРУТОНА СИНДРОМ
Первые симптомы заболевания проявляются, как правило, в возрасте менее 1 года,

Слайд 25СИНДРОМ ЛУИ БАР
ИДС проявляющееся в виде одной из форм атаксии (нарушения координации
СИНДРОМ ЛУИ БАР
ИДС проявляющееся в виде одной из форм атаксии (нарушения координации
Слайд 26ЛУИ-БАР СИНДРОМ
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у
ЛУИ-БАР СИНДРОМ
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у

Слайд 27ИОВА СИНДРОМ
(по имени библейского персонажа, пораженного «проказою лютою от подошвы ноги его
ИОВА СИНДРОМ
(по имени библейского персонажа, пораженного «проказою лютою от подошвы ноги его

Слайд 28ОБЩИЕ ПРИНЦИПЫ
При тотальных и субтотальных дефицитах антителопродукции вакцинация неэффективна
Дефект клеточного звена –
ОБЩИЕ ПРИНЦИПЫ
При тотальных и субтотальных дефицитах антителопродукции вакцинация неэффективна
Дефект клеточного звена –

Слайд 29ПРИОБРЕТЕННЫЕ ИДС
Обусловлены качественным и количественным голоданием (недостатком белков, витаминов, микроэлементов, Fe, Zn,
ПРИОБРЕТЕННЫЕ ИДС
Обусловлены качественным и количественным голоданием (недостатком белков, витаминов, микроэлементов, Fe, Zn,

Слайд 30ПЕРЕЧЕНЬ ОСНОВНЫХ ЗАБОЛЕВАНИЙ, СОПРОВОЖДАЮЩИХСЯ ВТОРИЧНЫМ ИММУНОДЕФИЦИТОМ (ВОЗ)
Инфекционные заболевания:
Протозойные и глистные
ПЕРЕЧЕНЬ ОСНОВНЫХ ЗАБОЛЕВАНИЙ, СОПРОВОЖДАЮЩИХСЯ ВТОРИЧНЫМ ИММУНОДЕФИЦИТОМ (ВОЗ)
Инфекционные заболевания:
Протозойные и глистные

Слайд 31Нарушения питания: истощение, кахексия, нарушения кишечного всасывания и др.
Экзогенные и эндогенные
Нарушения питания: истощение, кахексия, нарушения кишечного всасывания и др.
Экзогенные и эндогенные

Слайд 32Действие различных видов излучения, особенно ионизирующей радиации.
Сильные, длительные стрессорные воздействия.
Действие
Действие различных видов излучения, особенно ионизирующей радиации.
Сильные, длительные стрессорные воздействия.
Действие

Слайд 33КЛИНИЧЕСКИЕ ПРИЗНАКИ ИММУНОДЕФИЦИТНОГО СОСТОЯНИЯ:
Частые обострения хронических воспалительных заболеваний разной этиологии
Частые обострения герпетической
КЛИНИЧЕСКИЕ ПРИЗНАКИ ИММУНОДЕФИЦИТНОГО СОСТОЯНИЯ:
Частые обострения хронических воспалительных заболеваний разной этиологии
Частые обострения герпетической

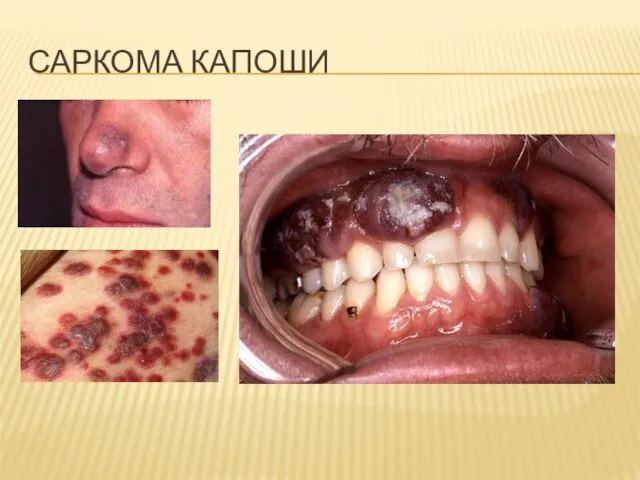
Слайд 36САРКОМА КАПОШИ
САРКОМА КАПОШИ
Слайд 37ГЛАВНЫЙ КОМПЛЕКС ГИСТОСОВМЕСТИМОСТИ – ЭТО ГРУППА ГЕНОВ И КОДИРУЕМЫХ ИМИ АНТИГЕНОВ КЛЕТОЧНОЙ
ГЛАВНЫЙ КОМПЛЕКС ГИСТОСОВМЕСТИМОСТИ – ЭТО ГРУППА ГЕНОВ И КОДИРУЕМЫХ ИМИ АНТИГЕНОВ КЛЕТОЧНОЙ

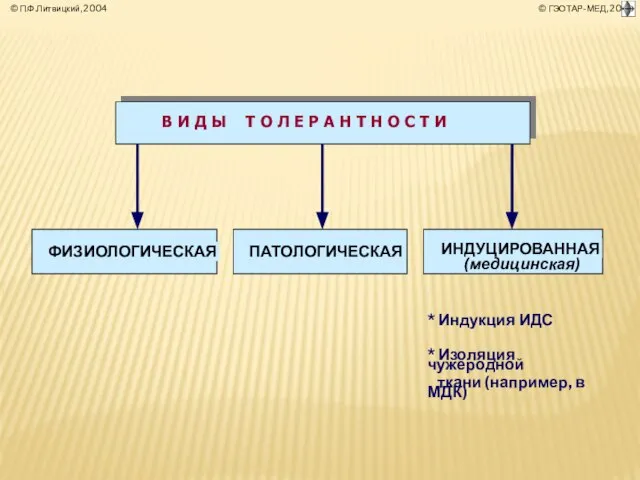
Слайд 38ПАТОЛОГИЧЕСКАЯ Т О Л Е Р А Н Т Н О С
ПАТОЛОГИЧЕСКАЯ Т О Л Е Р А Н Т Н О С


Слайд 43РТПХ
РАНТ-болезнь в эксперименте.
РТПХ
РАНТ-болезнь в эксперименте.



Слайд 46ФОРМИРОВАНИЕ ТОЛЕРАНТНОСТИ К ТРАНСПЛАНТАТУ
Неспецифические методы:
1. Подавление иммунологической реактивности организма реципиента с
ФОРМИРОВАНИЕ ТОЛЕРАНТНОСТИ К ТРАНСПЛАНТАТУ
Неспецифические методы:
1. Подавление иммунологической реактивности организма реципиента с

Слайд 47ПАТОГЕНЕЗ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ
1) нарушение физиологической изоляции органов и тканей, по отношению к которым
ПАТОГЕНЕЗ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ
1) нарушение физиологической изоляции органов и тканей, по отношению к которым

Слайд 48МИАСТЕНИЯ ГРАВИС
МИАСТЕНИЯ ГРАВИС

Слайд 49СИНДРОМ ДРЕССЛЕРА
Постинфарктное осложнение
СИНДРОМ ДРЕССЛЕРА
Постинфарктное осложнение
 Склериты. Виды склеритов
Склериты. Виды склеритов Filariasis
Filariasis Этиология и патогенез атеросклроза
Этиология и патогенез атеросклроза Заболевания, передающиеся с водой
Заболевания, передающиеся с водой Қанат – таңдай түйінінің невралгиясы
Қанат – таңдай түйінінің невралгиясы Рестриктивная кардиомиопатия
Рестриктивная кардиомиопатия Осторожно! Вирус Covid-19!
Осторожно! Вирус Covid-19! Артериальная гипертония
Артериальная гипертония Пойдете ли Вы на работу, заболев гриппом?
Пойдете ли Вы на работу, заболев гриппом? Патологии зрительного и глазодвигательных нервов
Патологии зрительного и глазодвигательных нервов Профилактика наркозависимости
Профилактика наркозависимости атипичные пневмонии узбекча
атипичные пневмонии узбекча ХОБЛ: незаметный убийца
ХОБЛ: незаметный убийца Методы идентификации клеточных элементов в моче: суправитальная окраска препаратов осадка мочи, подсчёт уролейкограммы
Методы идентификации клеточных элементов в моче: суправитальная окраска препаратов осадка мочи, подсчёт уролейкограммы Решение клинических кейсов диспансерное наблюдение детей с заболеваниями органов дыхания
Решение клинических кейсов диспансерное наблюдение детей с заболеваниями органов дыхания Анестизиология. Анафилактикалық шок
Анестизиология. Анафилактикалық шок Limfoscyntygrafia
Limfoscyntygrafia Особенности вирусного гепатита (A, B, C, D, E, F, G)
Особенности вирусного гепатита (A, B, C, D, E, F, G) Манчестерская операция /Операция Дональда, усовершенствованная Фозергиллом
Манчестерская операция /Операция Дональда, усовершенствованная Фозергиллом Атеросклероз. Нормальная интима аорты
Атеросклероз. Нормальная интима аорты The immediate (early phase) allergic reaction in the nose
The immediate (early phase) allergic reaction in the nose Урологические и андрологические заболевания при сахарном диабете
Урологические и андрологические заболевания при сахарном диабете Сифилис
Сифилис Оказание первой помощи при кровотечениях
Оказание первой помощи при кровотечениях Европейская медицина XVIII – XIX вв. Развитие медико-биологических наук
Европейская медицина XVIII – XIX вв. Развитие медико-биологических наук Диспансеризация детского населения. (Лекция 4)
Диспансеризация детского населения. (Лекция 4) История внедрения и перспективы применения компьютерных технологий в современной медицинской науке и практике
История внедрения и перспективы применения компьютерных технологий в современной медицинской науке и практике Мозг и его болезни
Мозг и его болезни