- Энзимопатии

Содержание
- 2. Процессы обмена веществ в клетке находятся под контролем: Нервной и эндокринной регуляции, обеспечивающих согласование обменных процессов
- 3. -Полная блокада (выключение) синтеза фермента; -Снижения активности фермента; -Нарушения других систем или биохимических реакций, от которых
- 4. ВЫЯВЛЕНИЕ И ДИАГНОЗ Фенилкетонурия и галактоземия (нарушение углеводного обмена), могут быть определены путем анализа крови, взятой
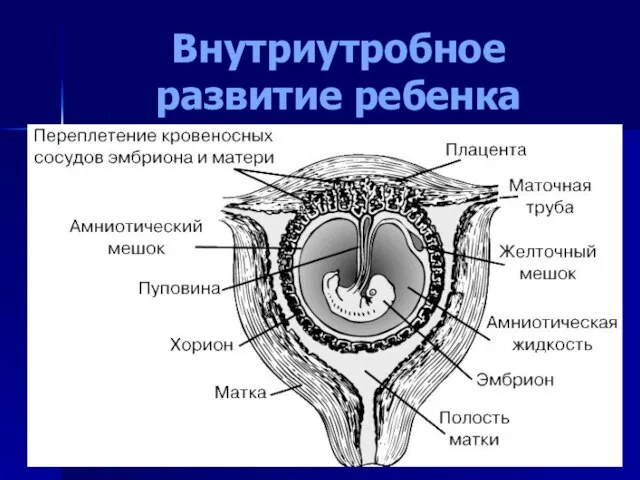
- 5. Внутриутробное развитие ребенка
- 7. Наследственные нарушения аминокислот (энзимопатии) - — группа заболеваний, обусловленных дефектами ферментов, участвующих в их обмене. Описано
- 8. Подавляющее большинство этих болезней наследуется аутосомно-рецессивно. Отдельные формы заболеваний передаются с Х-хромосомой. При большинстве нарушений аминокислотного
- 9. 1. Ограничение в диете белка и соответствующей аминокислоты. 2. Дополнительное назначение незаменимых аминокислот. 3. Назначение препаратов,
- 10. Примеры энзимопатий
- 11. Фенилкетонурия (ФКУ) Впервые описал A. Foiling в 1934 году. Частота встречаемости в России — 1:10000. ФКУ
- 12. Патогенез Поражение ЦНС вызывается недостаточностью фермента гидроксилазы-4-фенилаланина, управляющего превращением фенилаланина в тирозин. В результате этого концентрация
- 13. Гиперкинезы, нарушения мышечного тонуса и координации. 2. 25—50% больных страдают припадками. 3. Нарушения пигментации (светлый цвет
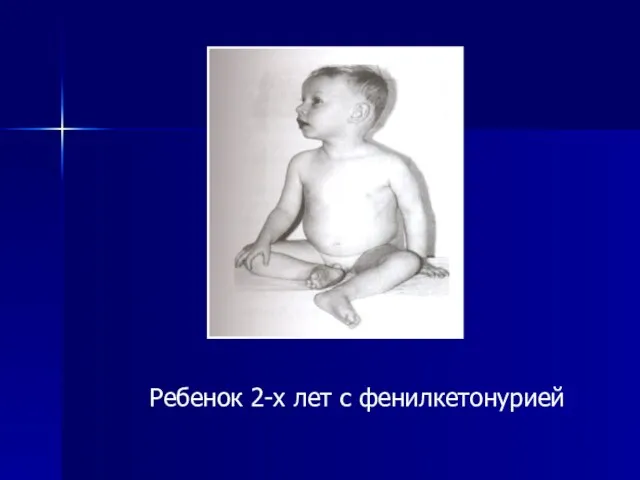
- 14. Ребенок 2-х лет с фенилкетонурией
- 15. Психическое развитие Отставание в психическом развитии становится заметным во втором полугодии жизни и прогрессирует в течение
- 16. Лечение Диета с резким ограничением фенилаланина с 2— 3 -месячного возраста и соблюдение ее в течение
- 17. ОРГАНИЧЕСКИЕ АЦИДЕМИИ, СОПРОВОЖДАЮЩИЕСЯ НАРУШЕНИЕМ НЕРВНО-ПСИХИЧЕСКОГО РАЗВИТИЯ Эти нарушения метаболизма органических кислот (продуктов обмена аминокислот, углеводов, липидов,
- 18. В 1967 году (Budd M. А. et al.) впервые была уточнена природа одной из ацидемий (изовалериановой).
- 19. Первичные симптомы: - респираторный и нейродистресс-синдромы, - припадки, - рвота, отказ от еды, нарушение стула, обезвоживание.
- 20. Вторичные нарушения: - отставание психического и моторного развития, -пирамидная симптоматика, -расстройства координации, -судороги. Особые симптомы отмечаются
- 21. Лечение ограничение белка; высокие дозах витаминов; дополнительное введение Л-карнитина и глицина.
- 22. Галактоземия Описана в 1908 году, однако дефект обмена, ее обуславливающий, был открыт лишь в 1956 году.
- 23. Патогенез Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не усваивается, а промежуточный продукт
- 24. Клиника Проявляется вскоре после рождения у ребенка: отказом от пищи, поносом, рвотой, непереносимостью голода, падением массы
- 25. Лечение Безмолочная диета
- 26. МУКОПОЛИСАХАРИДО3 1 Н (СИНДРОМ ГУРЛЕРА) Описан G. Gurler в 1919 году. Встречается с частотой — 1:
- 27. Клиника Проявляется на первом году жизни. Внешний вид больных — увеличенная голова, выдающиеся лобные бугры, почти
- 28. Умственная отсталость заметна уже в раннем возрасте. В последующем интеллектуальный дефект усугубляется, затем происходит потеря приобретенных
- 29. Патогенез Отложение мукополисахаридов в соединительной ткани печени, селезенки и других тканях. Накопление мукополисахаридов в хрящах нарушает
- 30. Болезнь Гирке Нарушение обмена веществ, характеризующееся накоплением избыточного количества гликогена в тканях организма. Связано с недостаточностью
- 31. Альбинизм При нормальном метаболизме фенилаланина и тирозина (обе аминокислоты связаны между собой в обмене) образуется кожный
- 32. Алкаптонурия Заболевание вызывается генетически обусловленной недостаточностью фермента, участвующего в метаболизме гомогентизиновой кислоты — промежуточного продукта обмена
- 33. Гиперхолестеринемия Неспособность организма разрушать холестерин и липопротеины низкой плотности (в составе которых он в основном находится)
- 35. Скачать презентацию

Слайд 3-Полная блокада (выключение) синтеза фермента;
-Снижения активности фермента;
-Нарушения других систем или
-Полная блокада (выключение) синтеза фермента; -Снижения активности фермента; -Нарушения других систем или

Слайд 4
ВЫЯВЛЕНИЕ И ДИАГНОЗ
Фенилкетонурия и галактоземия (нарушение углеводного обмена), могут быть определены путем анализа
ВЫЯВЛЕНИЕ И ДИАГНОЗ
Фенилкетонурия и галактоземия (нарушение углеводного обмена), могут быть определены путем анализа

Слайд 5Внутриутробное развитие ребенка
Внутриутробное развитие ребенка
Слайд 7 Наследственные нарушения аминокислот (энзимопатии) -
— группа заболеваний, обусловленных дефектами ферментов,
Наследственные нарушения аминокислот (энзимопатии) -
— группа заболеваний, обусловленных дефектами ферментов,

Слайд 8 Подавляющее большинство этих болезней наследуется аутосомно-рецессивно.
Отдельные формы заболеваний передаются с Х-хромосомой.
Подавляющее большинство этих болезней наследуется аутосомно-рецессивно. Отдельные формы заболеваний передаются с Х-хромосомой.

Слайд 91. Ограничение в диете белка и соответствующей аминокислоты.
2. Дополнительное назначение незаменимых аминокислот.
3.
1. Ограничение в диете белка и соответствующей аминокислоты. 2. Дополнительное назначение незаменимых аминокислот. 3.

Слайд 10Примеры энзимопатий

Слайд 11Фенилкетонурия
(ФКУ)
Впервые описал A. Foiling в 1934 году.
Частота встречаемости в России
Фенилкетонурия
(ФКУ)
Впервые описал A. Foiling в 1934 году.
Частота встречаемости в России

Слайд 12Патогенез
Поражение ЦНС вызывается недостаточностью фермента гидроксилазы-4-фенилаланина, управляющего превращением фенилаланина в
Патогенез
Поражение ЦНС вызывается недостаточностью фермента гидроксилазы-4-фенилаланина, управляющего превращением фенилаланина в

Слайд 13 Гиперкинезы, нарушения мышечного тонуса и координации.
2. 25—50% больных страдают припадками.
Гиперкинезы, нарушения мышечного тонуса и координации. 2. 25—50% больных страдают припадками.

Слайд 14
Ребенок 2-х лет с фенилкетонурией
Ребенок 2-х лет с фенилкетонурией
Слайд 15Психическое развитие
Отставание в психическом развитии становится заметным во втором полугодии жизни
Психическое развитие
Отставание в психическом развитии становится заметным во втором полугодии жизни

Слайд 16Лечение
Диета с резким ограничением фенилаланина с 2— 3 -месячного возраста и соблюдение
Лечение
Диета с резким ограничением фенилаланина с 2— 3 -месячного возраста и соблюдение

Слайд 17ОРГАНИЧЕСКИЕ АЦИДЕМИИ, СОПРОВОЖДАЮЩИЕСЯ НАРУШЕНИЕМ
НЕРВНО-ПСИХИЧЕСКОГО РАЗВИТИЯ
Эти нарушения метаболизма органических кислот
ОРГАНИЧЕСКИЕ АЦИДЕМИИ, СОПРОВОЖДАЮЩИЕСЯ НАРУШЕНИЕМ
НЕРВНО-ПСИХИЧЕСКОГО РАЗВИТИЯ
Эти нарушения метаболизма органических кислот

Слайд 18 В 1967 году (Budd M. А. et al.) впервые была уточнена природа
В 1967 году (Budd M. А. et al.) впервые была уточнена природа

Слайд 19
Первичные симптомы:
- респираторный и нейродистресс-синдромы,
- припадки,
- рвота, отказ от еды, нарушение
Первичные симптомы: - респираторный и нейродистресс-синдромы, - припадки, - рвота, отказ от еды, нарушение

Слайд 20
Вторичные нарушения:
- отставание психического и моторного развития,
-пирамидная симптоматика,
-расстройства координации,
Вторичные нарушения: - отставание психического и моторного развития, -пирамидная симптоматика, -расстройства координации,

Слайд 21Лечение
ограничение белка;
высокие дозах витаминов;
дополнительное введение Л-карнитина и глицина.
Лечение
ограничение белка;
высокие дозах витаминов;
дополнительное введение Л-карнитина и глицина.

Слайд 22Галактоземия
Описана в 1908 году,
однако дефект обмена,
ее обуславливающий,
был открыт лишь
Галактоземия
Описана в 1908 году,
однако дефект обмена,
ее обуславливающий,
был открыт лишь

Слайд 23Патогенез
Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не
Патогенез
Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не

Слайд 24Клиника
Проявляется вскоре после рождения у ребенка:
отказом от пищи, поносом, рвотой, непереносимостью голода,
Клиника
Проявляется вскоре после рождения у ребенка:
отказом от пищи, поносом, рвотой, непереносимостью голода,

Слайд 25Лечение
Безмолочная диета
Лечение
Безмолочная диета

Слайд 26МУКОПОЛИСАХАРИДО3 1 Н (СИНДРОМ ГУРЛЕРА)
Описан G. Gurler в 1919 году.
Встречается с
МУКОПОЛИСАХАРИДО3 1 Н (СИНДРОМ ГУРЛЕРА)
Описан G. Gurler в 1919 году.
Встречается с

Слайд 27
Клиника
Проявляется на первом году жизни.
Внешний вид больных — увеличенная голова, выдающиеся лобные
Клиника
Проявляется на первом году жизни.
Внешний вид больных — увеличенная голова, выдающиеся лобные

Слайд 28 Умственная отсталость заметна уже в раннем возрасте.
В последующем интеллектуальный дефект усугубляется,
Умственная отсталость заметна уже в раннем возрасте. В последующем интеллектуальный дефект усугубляется,

Слайд 29Патогенез
Отложение мукополисахаридов в соединительной ткани печени, селезенки и других тканях. Накопление мукополисахаридов
Патогенез
Отложение мукополисахаридов в соединительной ткани печени, селезенки и других тканях. Накопление мукополисахаридов

Слайд 30
Болезнь Гирке
Нарушение обмена веществ, характеризующееся накоплением избыточного количества гликогена в тканях организма.
Болезнь Гирке
Нарушение обмена веществ, характеризующееся накоплением избыточного количества гликогена в тканях организма.

Слайд 31Альбинизм
При нормальном метаболизме фенилаланина и тирозина (обе аминокислоты связаны между собой в обмене) образуется
Альбинизм
При нормальном метаболизме фенилаланина и тирозина (обе аминокислоты связаны между собой в обмене) образуется

Слайд 32
Алкаптонурия
Заболевание вызывается генетически обусловленной недостаточностью фермента, участвующего в метаболизме гомогентизиновой кислоты — промежуточного
Алкаптонурия
Заболевание вызывается генетически обусловленной недостаточностью фермента, участвующего в метаболизме гомогентизиновой кислоты — промежуточного

Слайд 33Гиперхолестеринемия
Неспособность организма разрушать холестерин и липопротеины низкой плотности (в составе которых он в основном находится)
Гиперхолестеринемия
Неспособность организма разрушать холестерин и липопротеины низкой плотности (в составе которых он в основном находится)

 Наконец летит!
Наконец летит! Структура дерева целей ООО ЖК курортный
Структура дерева целей ООО ЖК курортный Задачи на вычисление импульса тела.
Задачи на вычисление импульса тела. Родительское собрание
Родительское собрание Александр Невский
Александр Невский Уровни организации жизни
Уровни организации жизни Влияние сквернословия на здоровье человека
Влияние сквернословия на здоровье человека Организация производства натуральных эфирных масел из экологически чистого сырья
Организация производства натуральных эфирных масел из экологически чистого сырья MGQ_Lore_1
MGQ_Lore_1 Webinar requirement form
Webinar requirement form Качество образования – качество жизни
Качество образования – качество жизни Введение во храм Пресвятой Богородицы
Введение во храм Пресвятой Богородицы Первый межрегиональный конкурс Университетская книга – Золотое кольцо
Первый межрегиональный конкурс Университетская книга – Золотое кольцо Соль Мертвого моря Иорданская Грязь Мертвого моря минеральная «Крымская» розовая соль
Соль Мертвого моря Иорданская Грязь Мертвого моря минеральная «Крымская» розовая соль Индивидуальное здоровье, его физическая, духовная и социальная сущность
Индивидуальное здоровье, его физическая, духовная и социальная сущность Выбор действий при решении задач
Выбор действий при решении задач Презентация на тему Комфорт обучающихся на занятиях в кружках
Презентация на тему Комфорт обучающихся на занятиях в кружках Презентация на тему Принципы гражданского и арбитражного процесса
Презентация на тему Принципы гражданского и арбитражного процесса Растения хищники
Растения хищники Фиксирование тематик обращений
Фиксирование тематик обращений Познакомьтесь, это мы!
Познакомьтесь, это мы! Юдникова 26.10
Юдникова 26.10 La Navidad en España
La Navidad en España Части вдохновляющей одежды
Части вдохновляющей одежды Матрешки. Загорская матрёшка
Матрешки. Загорская матрёшка Реверсивные устройства
Реверсивные устройства Правовое государство
Правовое государство Казанский государственный медицинский университет. Кафедра офтальмологии. Электронное учебное пособие
Казанский государственный медицинский университет. Кафедра офтальмологии. Электронное учебное пособие