- Молекула в расчетных методах

Содержание
- 2. Индексы i, j относятся к электронам; a, b – к ядрам; M и m – массы
- 3. Разделить действия гамильтониана Ĥ на ядерную и электронную части точной Ψ(r,R) волновой функции нельзя. Это можно
- 4. ΨЭ({r,R}) – удовлетворяет уравнению: Ĥэ ΨЭ({r,R}) = EэΨЭ({r,R}), где ΨЯ({R}) – удовлетворяет уравнению:
- 5. Полная энергия молекул в приближении Борна Оппенгеймера есть: Е = ЕЭ + ТЯ, ТЯ – колебательно–вращательная
- 6. Полная энергия системы Хартри-Фока для молекулы: При учете приближения Борна-Оппенгеймера уравнения Хартри-Фока для молекулы можно решить
- 7. Приближение МО ЛКАО. Уравнения Рутана. Двигаясь по молекуле (состоящей из m атомов), каждый электрон попадает под
- 8. Другими словами - Теория метода Молекулярных Орбиталей исходит из предположения, что первоначально сближаются только ядра вместе
- 9. Обобщенное уравнение на собственные значения называется уравнением Рутана: F- матрица оператора Фока, S- матрица перекрытия для
- 10. Различают метод Харти-Фока-Рутана для систем с замкнутыми электронными оболочками и с открытыми электронными оболочками. Для замкнутых
- 11. Однако из-за снятия указанного фундаментального ограничения на электронную волновую функцию, ее смысл в НХФ часто подвергается
- 12. Теория возмущения: Приближения Меллера-Плессета: MP2, MP3, MP4 Уравнение Шредингера (уравнение 1, Лекция 1) имеет вид: Н
- 13. Конфигурационное взаимодействие Волновая функция (Ψкв) ищется вариационным методом в виде линейной комбинации молекулярных орбиталей Ψк, которые
- 14. Энергии диссоциации (ккал/моль) молекул, рассчитанные различными методами
- 15. Канонические и локализованные молекулярные орбитали Молекулярные орбитали ϕi определяются как собственные функции некоторого одноэлектронного гамильтониана F
- 16. В качестве примера – молекула метана: CH4 ( точечная симметрия Td) Канонические МО: ϕ1 = a(2sC)
- 17. Указанные канонические МО можно преобразовать в локализованные МО : fi = c(hiC) + d(1sHi) + (a-c)(2sC)
- 18. Теория метода Молекулярных Орбиталей исходит из предположения, что первоначально сближаются только ядра вместе с внутренними оболочками.
- 19. Примеры: метод ВС метод ЛКАО МО e1 e2 атомы H A B Ψ1 = ΨA(e1) *
- 20. Метод ВС предполагает, что атомы в молекулах сохраняют во многом свою индивидуальность, а эффект химической связи
- 21. Какой метод лучше? Метод ВС дает более наглядное представление о химических связях Метод МО не требует
- 23. Скачать презентацию

Слайд 3Разделить действия гамильтониана Ĥ на ядерную и электронную части точной Ψ(r,R) волновой
Разделить действия гамильтониана Ĥ на ядерную и электронную части точной Ψ(r,R) волновой

Слайд 4ΨЭ({r,R}) – удовлетворяет уравнению:
Ĥэ ΨЭ({r,R}) = EэΨЭ({r,R}), где
ΨЯ({R}) – удовлетворяет уравнению:
ΨЭ({r,R}) – удовлетворяет уравнению:
Ĥэ ΨЭ({r,R}) = EэΨЭ({r,R}), где
ΨЯ({R}) – удовлетворяет уравнению:

Слайд 5Полная энергия молекул в приближении Борна Оппенгеймера есть:
Е = ЕЭ +
Полная энергия молекул в приближении Борна Оппенгеймера есть:
Е = ЕЭ +

Слайд 6Полная энергия системы Хартри-Фока для молекулы:
При учете приближения Борна-Оппенгеймера уравнения Хартри-Фока для
Полная энергия системы Хартри-Фока для молекулы:
При учете приближения Борна-Оппенгеймера уравнения Хартри-Фока для

Слайд 7Приближение МО ЛКАО. Уравнения Рутана.
Двигаясь по молекуле (состоящей из m атомов),
Приближение МО ЛКАО. Уравнения Рутана.
Двигаясь по молекуле (состоящей из m атомов),

Слайд 8Другими словами - Теория метода Молекулярных Орбиталей исходит из предположения, что первоначально
Другими словами - Теория метода Молекулярных Орбиталей исходит из предположения, что первоначально

Слайд 9Обобщенное уравнение на собственные значения называется уравнением Рутана:
F- матрица оператора Фока, S-
Обобщенное уравнение на собственные значения называется уравнением Рутана:
F- матрица оператора Фока, S-

Слайд 10Различают метод Харти-Фока-Рутана для систем с замкнутыми электронными оболочками и с открытыми
Различают метод Харти-Фока-Рутана для систем с замкнутыми электронными оболочками и с открытыми
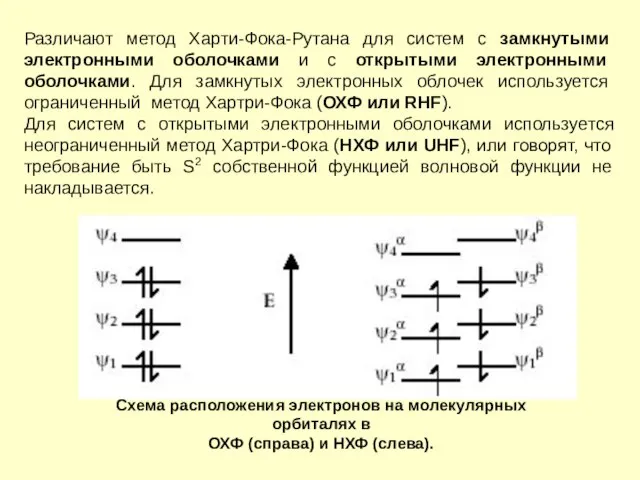
Слайд 11Однако из-за снятия указанного фундаментального ограничения на электронную волновую функцию, ее смысл
Однако из-за снятия указанного фундаментального ограничения на электронную волновую функцию, ее смысл

Слайд 12Теория возмущения:
Приближения Меллера-Плессета: MP2, MP3, MP4
Уравнение Шредингера (уравнение 1, Лекция
Теория возмущения:
Приближения Меллера-Плессета: MP2, MP3, MP4
Уравнение Шредингера (уравнение 1, Лекция
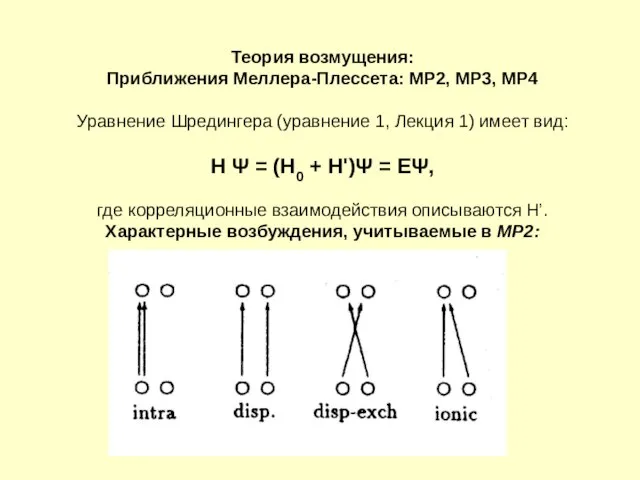
Слайд 13Конфигурационное взаимодействие
Волновая функция (Ψкв) ищется вариационным методом в виде линейной комбинации молекулярных
Конфигурационное взаимодействие
Волновая функция (Ψкв) ищется вариационным методом в виде линейной комбинации молекулярных

Слайд 14Энергии диссоциации (ккал/моль) молекул,
рассчитанные различными методами
Энергии диссоциации (ккал/моль) молекул,
рассчитанные различными методами
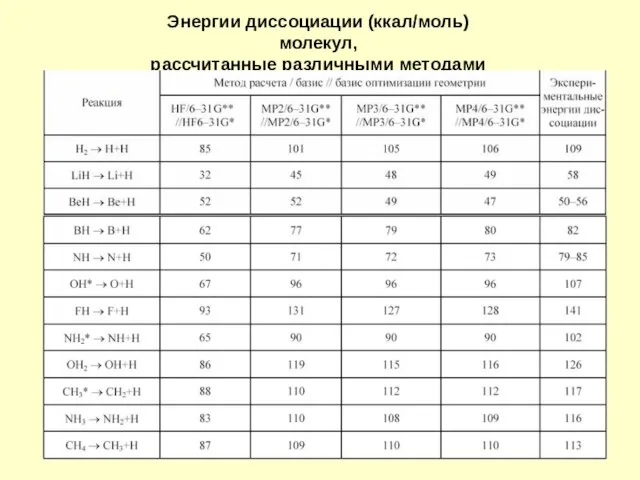
Слайд 15 Канонические и локализованные молекулярные орбитали
Молекулярные орбитали ϕi определяются как собственные функции
Канонические и локализованные молекулярные орбитали
Молекулярные орбитали ϕi определяются как собственные функции

Слайд 16 В качестве примера – молекула метана: CH4
( точечная симметрия
В качестве примера – молекула метана: CH4
( точечная симметрия

Слайд 17Указанные канонические МО можно преобразовать
в локализованные МО :
fi = c(hiC)
Указанные канонические МО можно преобразовать
в локализованные МО :
fi = c(hiC)

Слайд 18Теория метода Молекулярных Орбиталей исходит из предположения, что первоначально сближаются только ядра
Теория метода Молекулярных Орбиталей исходит из предположения, что первоначально сближаются только ядра

Слайд 19Примеры: метод ВС метод ЛКАО МО
e1 e2
атомы H A
Примеры: метод ВС метод ЛКАО МО
e1 e2
атомы H A

Слайд 20 Метод ВС предполагает, что атомы в молекулах сохраняют во многом свою индивидуальность,
Метод ВС предполагает, что атомы в молекулах сохраняют во многом свою индивидуальность,

Слайд 21Какой метод лучше?
Метод ВС дает более наглядное представление о химических связях
Метод МО
Какой метод лучше?
Метод ВС дает более наглядное представление о химических связях
Метод МО

 Родительское собрание«Трудности подросткового возраста»
Родительское собрание«Трудности подросткового возраста» Приказ Минфина № 126 н. Как не подарить государству 15 (20) %
Приказ Минфина № 126 н. Как не подарить государству 15 (20) % Сведения об имени числительном
Сведения об имени числительном Презентация
Презентация Уфимский колледж индустрии питания и сервиса г. Салават. Профессии и специальности
Уфимский колледж индустрии питания и сервиса г. Салават. Профессии и специальности МЕНЕДЖМЕНТ
МЕНЕДЖМЕНТ Алгоритмы
Алгоритмы Выручка по договорам с покупателями
Выручка по договорам с покупателями Библейская тема в творчестве Л.да Винчи, Рембрандта А.Иванова, М.Нестерова
Библейская тема в творчестве Л.да Винчи, Рембрандта А.Иванова, М.Нестерова Исполнительная власть. Местные представительные и исполнительные органы
Исполнительная власть. Местные представительные и исполнительные органы Влияние физических упражнений на организм человека
Влияние физических упражнений на организм человека Где и как искать патентную информацию?
Где и как искать патентную информацию? Экономика
Экономика Подготовка к собеседованию
Подготовка к собеседованию Здоровая семья – здоровый ребёнок
Здоровая семья – здоровый ребёнок Европейское искусство XIX века. Портрет
Европейское искусство XIX века. Портрет Трагическое в искусстве
Трагическое в искусстве Открытие йода
Открытие йода Библиотека МКОУ СОШ с.Онот
Библиотека МКОУ СОШ с.Онот Особенности лирики Ксении Некрасовой
Особенности лирики Ксении Некрасовой «ЭКСПРЕСС-БЮДЖЕТИРОВАНИЕ»ЭФФЕКТИВНОЕ БЮДЖЕТНОЕ УПРАВЛЕНИЕ
«ЭКСПРЕСС-БЮДЖЕТИРОВАНИЕ»ЭФФЕКТИВНОЕ БЮДЖЕТНОЕ УПРАВЛЕНИЕ Психодрама. Цели и задачи психодрамы
Психодрама. Цели и задачи психодрамы Project work of Business English Language Female managers
Project work of Business English Language Female managers Моделирование эволюции
Моделирование эволюции Организация системы деятельности библиотеки общеобразовательной организации
Организация системы деятельности библиотеки общеобразовательной организации Медвежий промысел
Медвежий промысел Интерактивный Годовой отчет в 2011,
Интерактивный Годовой отчет в 2011, Содержание личных прав, свобод.
Содержание личных прав, свобод.