- Мукополисахаридоз типа I-Н (синдром Гурлер)

Содержание
- 2. Мукополисахаридоз типа I-Н (синдром Гурлер) - - наследственные заболевания обмена веществ, относящиеся к группе лизосомных болезней
- 3. Что провоцирует Мукополисахаридоз типа I-Н (синдром Гурлер): - аутосомно-рецессивное заболевание, обусловленное мутациями в структурном гене лизосомного
- 4. Патогенез (что происходит?) во время Мукополисахаридоза типа I-Н (синдрома Гурлера) Патоморфологическая картина: наблюдается утолщение костей черепа
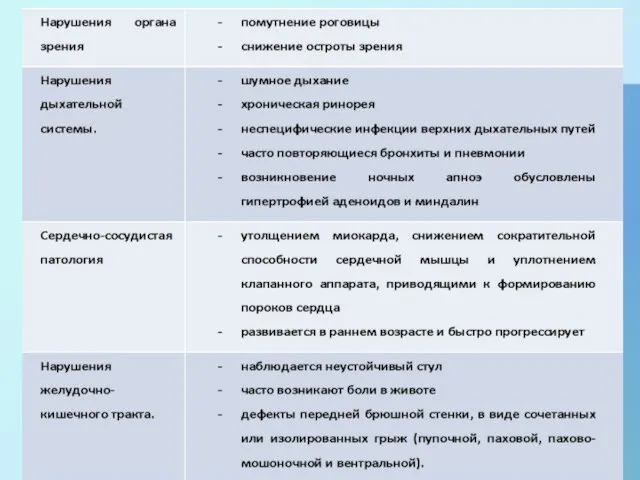
- 5. Нарушения при МПС I-H:
- 8. Особенности фенотипа. Характерны изменения черт лица по типу «гаргоилизма», которые становятся очевидными к концу первого года
- 9. Клиническая картина МПС I-H: Первые клинические признаки заболевания появляются на первом году жизни. В ряде случаев,
- 10. Клиническая диагностика В ряде случаев, выраженный клинический полиморфизм МПС I приводит к ошибочной диагностике, что в
- 11. Диагностика Мукополисахаридоза типа I-Н (синдрома Гурлера): Подтверждающая биохимическая диагностика МПС I заключается в определении уровня экскреции
- 12. Лечение Мукополисахаридоза типа I-Н (синдрома Гурлера): Заменительная терапия. Трансплантация стволовых клеток. Хирургическая коррекция глаукомы, скелетных аномалий,
- 14. Скачать презентацию
Слайд 2Мукополисахаридоз типа I-Н (синдром Гурлер) -
- наследственные заболевания обмена веществ, относящиеся
Мукополисахаридоз типа I-Н (синдром Гурлер) -
- наследственные заболевания обмена веществ, относящиеся

Слайд 3Что провоцирует Мукополисахаридоз типа I-Н (синдром Гурлер):
- аутосомно-рецессивное заболевание, обусловленное мутациями в
Что провоцирует Мукополисахаридоз типа I-Н (синдром Гурлер):
- аутосомно-рецессивное заболевание, обусловленное мутациями в

Слайд 4Патогенез (что происходит?) во время Мукополисахаридоза типа I-Н (синдрома Гурлера)
Патоморфологическая картина:
наблюдается утолщение
Патогенез (что происходит?) во время Мукополисахаридоза типа I-Н (синдрома Гурлера)
Патоморфологическая картина:
наблюдается утолщение

Слайд 5Нарушения при МПС I-H:
Нарушения при МПС I-H:


Слайд 8Особенности фенотипа.
Характерны изменения черт лица по типу «гаргоилизма», которые становятся очевидными
Особенности фенотипа.
Характерны изменения черт лица по типу «гаргоилизма», которые становятся очевидными

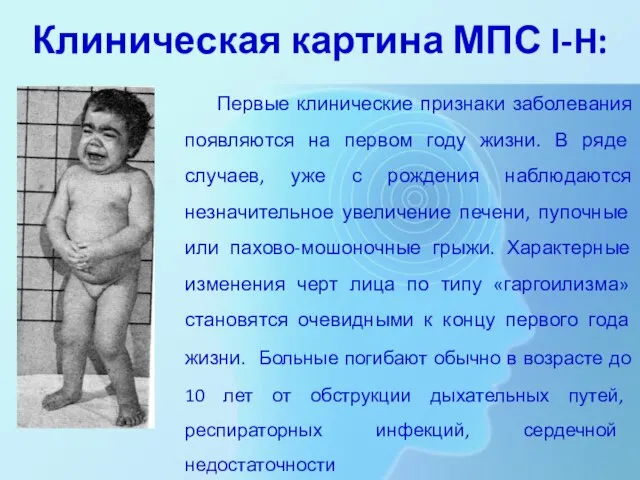
Слайд 9Клиническая картина МПС I-H:
Первые клинические признаки заболевания появляются на первом году
Клиническая картина МПС I-H:
Первые клинические признаки заболевания появляются на первом году
Слайд 10Клиническая диагностика
В ряде случаев, выраженный клинический полиморфизм МПС I приводит к
Клиническая диагностика
В ряде случаев, выраженный клинический полиморфизм МПС I приводит к

Слайд 11Диагностика Мукополисахаридоза типа I-Н (синдрома Гурлера):
Подтверждающая биохимическая диагностика МПС I заключается в
Диагностика Мукополисахаридоза типа I-Н (синдрома Гурлера):
Подтверждающая биохимическая диагностика МПС I заключается в

Слайд 12Лечение Мукополисахаридоза типа I-Н (синдрома Гурлера):
Заменительная терапия.
Трансплантация стволовых клеток.
Хирургическая коррекция глаукомы, скелетных
Лечение Мукополисахаридоза типа I-Н (синдрома Гурлера):
Заменительная терапия.
Трансплантация стволовых клеток.
Хирургическая коррекция глаукомы, скелетных

 Ошибки судов при проведении досудебной подготовки
Ошибки судов при проведении досудебной подготовки Лот 22, г. Хабаровск, ул. Сысоева, 21, кв. 65
Лот 22, г. Хабаровск, ул. Сысоева, 21, кв. 65 Предметы марийской национальной одежды в фондах Калтасинского музея
Предметы марийской национальной одежды в фондах Калтасинского музея Современные требования к организации уроков как эффективное средство повышения качества образования
Современные требования к организации уроков как эффективное средство повышения качества образования Услуги ООО Машаудит
Услуги ООО Машаудит Презентация на тему Обучение грамоте 1 класс "Знакомство со звуком а, буквой А"
Презентация на тему Обучение грамоте 1 класс "Знакомство со звуком а, буквой А" Ясько Роман Викторович
Ясько Роман Викторович Введение в язык программирования. Технологии программирования
Введение в язык программирования. Технологии программирования Мотивы народной песни в лирике Н.А. Некрасова
Мотивы народной песни в лирике Н.А. Некрасова Урок хлеба
Урок хлеба Подготовка проекта к печати
Подготовка проекта к печати Война и мир
Война и мир Орфоэпические нормы (произношение согласных звуков, ударение)
Орфоэпические нормы (произношение согласных звуков, ударение) Акушерские кровотечения
Акушерские кровотечения Тест «Внутренняя и внешняя политика Николая I»
Тест «Внутренняя и внешняя политика Николая I» Программный комплекс учета и сбыта электрической энергии BreeZe, BreeZeLaw
Программный комплекс учета и сбыта электрической энергии BreeZe, BreeZeLaw ГАЗЕТА «РОССИЙСКОЕ СТРАХОВАНИЕ»ДЛЯ ПАРТНЕРОВ
ГАЗЕТА «РОССИЙСКОЕ СТРАХОВАНИЕ»ДЛЯ ПАРТНЕРОВ Технические и технологические вопросы производства российских светодиодов
Технические и технологические вопросы производства российских светодиодов Зачётная работа для МФК по курсу Основы предпринимательской деятельности. Туризм
Зачётная работа для МФК по курсу Основы предпринимательской деятельности. Туризм Клуб Путешественников - кафе
Клуб Путешественников - кафе ЗАДАЧИ РЕАЛИЗАЦИИ ПРОЕКТОВ ПОВЫШЕНИЯ НАДЕЖНОСТИ РАСПРЕДЕЛИТЕЛЬНЫХ ЭЛЕКТРИЧЕСКИХ СЕТЕЙ ЗА СЧЕТ НОРМАЛИЗАЦИИ ПОТОКОВВ РЕАКТИВНОЙ
ЗАДАЧИ РЕАЛИЗАЦИИ ПРОЕКТОВ ПОВЫШЕНИЯ НАДЕЖНОСТИ РАСПРЕДЕЛИТЕЛЬНЫХ ЭЛЕКТРИЧЕСКИХ СЕТЕЙ ЗА СЧЕТ НОРМАЛИЗАЦИИ ПОТОКОВВ РЕАКТИВНОЙ Государственное дошкольное образовательное учреждение комбинированного вида детский сад № 128 Невского района
Государственное дошкольное образовательное учреждение комбинированного вида детский сад № 128 Невского района ОСТЕОПОРОЗ
ОСТЕОПОРОЗ Серебряный век. Течения в современной русской прозе и поэзии
Серебряный век. Течения в современной русской прозе и поэзии ПУБЛИЧНЫЙ ДОКЛАД Муниципального автономного общеобразовательного учреждения Беломорского муниципального района «Беломорская
ПУБЛИЧНЫЙ ДОКЛАД Муниципального автономного общеобразовательного учреждения Беломорского муниципального района «Беломорская  Российская Академия наук Издательский комплекс "Наука" Издательство Академкнига/Учебник
Российская Академия наук Издательский комплекс "Наука" Издательство Академкнига/Учебник УЧЕБНЫЙ ЦЕНТР «Довузовское и дополнительное образование»
УЧЕБНЫЙ ЦЕНТР «Довузовское и дополнительное образование» Роль финансов в кругообороте капитала предприятия
Роль финансов в кругообороте капитала предприятия