- Наследственные эндокринопатии

Содержание
- 2. Наследственные болезни эндокринной системы обусловлены мутациями в генах гормонов, их рецепторов, а также ферментов биосинтеза и
- 3. Так, различные синдромы гиперальдостеронизма и гипоальдостеронизма связаны с избыточной продукцией или дефицитом альдостерона
- 4. Наследственная недостаточность гормона роста ассоциирована с различными вариантами карликовости, или нанизма. Нарушения тиреоидного обмена являются причиной
- 5. Аутоиммунное поражение островкового аппарата поджелудочной железы, а также инсулинорезистентность инсулинозависимых тканей приводят к нарушениям всех видов
- 6. В этиологии большинства случаев сахарного диабета участвуют как средовые, так и генетические факторы, хотя существуют и
- 7. Наследственный альдостеронизм
- 8. Нарушения водно-солевого обмена являются ведущими клиническими проявлениями различных наследственных типов альдостеронизма. Аутосомно-рецессивный врожденный гипоальдостеронизм, обусловлен мутациями
- 9. Этот ген кодирует полипептид 2 субсемейства 11b митохондриального цитохрома P-450, или альдостеронсинтетазу — фермент, катализирующий конечный
- 11. Как многие другие адреналовые цитохромы P-450, альдостеронсинтетаза обладает множественной ферментативной активностью. В клубочковой зоне надпочечников фермент
- 12. Мутации, затрагивающие разные активности фермента, приводят к двум аллельным вариантам заболевания с недостаточностью КМОI или КМОII
- 13. При биохимическом исследовании выявляются гипонатриемия, гиперкалиемия и ацидоз. При этих заболеваниях, вызывающих изолированную недостаточность минералокортикоидов, биосинтез
- 14. Недостаточность КМОI и КМОII ведет к накоплению прямых и отдаленных субстратов блокированных реакций: дезоксикортикостерона, кортикостерона и
- 15. При первом варианте с недостаточностью КМОI болезнь проявляется в неонатальном периоде в виде частого срыгивания, рвоты,
- 16. При втором варианте заболевания адреналовые кризы с выраженной потерей натрия и воды и увеличением концентрации калия
- 17. Наследственный гиперальдостеронизм является гетерогенной группой заболеваний. Аутосомно-доминантный семейный гиперальдостеронизм I типа, или глюкокортикоид-подавляемый гиперальдостеронизм обусловлен микроструктурными
- 18. Чаще этот тип гиперальдостеронизма рассматривается к одна из моногенных формам артериальной гипертензии - дексаметазон-чувствительная гипертония
- 19. Аутосомно-доминантный тип III заболевания обусловлен мутациями в гене KCNJ5, кодирующем субъединицу 5 G-чувствительного калиевого канала семейства
- 20. Нарушения в работе других ионных каналов также могут приводить к различным формам альдостеронизма. Так, первичный альдостеронизм
- 21. Сопутствующими проявлениями заболевания являются артериальная гипертензия, легочная гипертензия и врожденные пороки сердца
- 22. Синдром Барттера является необычной формой вторичного гиперальдостеронизма, при котором гипертрофия и гиперплазия юкстагломерулярных клеток почечных нефронов
- 23. Болезнь развивается вследствие нарушения реабсорбции хлорида натрия в восходящем колене петли Генле, где в норме реабсорбируется
- 24. Ведущими клиническими проявлениями заболевания являются задержка роста, гиперактивность ренин-ангиотензиновой системы, гипокалиемия, увеличение продукции почечных простагландинов, гиперкальциурия
- 25. Наследственные формы синдрома Барттера – это гетерогенная группа аутосомно-рецессивных заболеваний, вызванных нарушением работы почечных ионных каналов.
- 26. Антенатальный тип 2 синдрома Барттера связан с мутациями в гене АТФ-чувствительного калиевого канала – KCNJ1. Тип
- 27. Причиной развития младенческого дигенного типа 4B синдрома Барттера, сочетающегося с нейросенсорной тугоухостью, является одновременное присутствие мутаций
- 28. Псевдогипоальдостеронизм I типа обусловлен снижением чувствительности канальцевого эпителия к альдостерону и нарушением реабсорбции натрия, приводящим к
- 29. Типичными для этой патологии являются инфекционные поражения дыхательных путей. Компенсация водно-солевого обмена, введение натрия и контроль
- 30. Наследственные формы заболевания генетически гетерогенны. Аутосомно-доминантная форма псевдогипоальдостеронизма типа IА с относительно мягким течением обусловлена мутациями
- 31. Причиной развития аутосомно-рецессивного псевдогипоальдостеронизма типа IВ являются нарушения работы эпителиальных натриевых каналов, обусловленные мутациями в генах
- 32. Псевдогипоальдостеронизм II типа, известный как синдром Гордона, клинически характеризуется гиперкалиемией, гиперхлоремией, метаболическим ацидозом, а также психическими
- 33. Наследственные формы заболевания также генетически гетерогенны. Два аутосомно-доминантных типа заболевания обусловлены мутациями в генах WNK4 и
- 34. Другим регулятором этого котранспортера является транскрипционный фактор kelch3, кодируемый геном KLHL3. Мутации в этом гене найдены
- 35. Еще один аутосомно-доминантный тип заболевания обусловлен мутациями в гене куллина 3 – CUL3. Куллины участвуют в
- 36. Таким образом, все генетические формы псевдогипоальдостеронизма II типа связаны с нарушением работы тиазид-чувствительного Na-Cl—котранспортера
- 37. Гипофизарный нанизм
- 38. Рост скелета и мягких тканей организма индуцируется гормоном роста (соматотропином), который синтезируется соматотропными клетками передней доли
- 39. После высвобождения из гипотоламуса биологически активный гормон роста связывается со своим трансмембранным рецептором, который димеризуется и
- 40. Соматомедины являются членами инсулинового семейства полипептидных факторов роста и выполняют роль аутокринных регуляторов клеточной пролиферации. Комплекс
- 41. Существует два гена гормона роста: нормальный — GH1, или GHN, и вариантный — GH2, или GHV,
- 42. В настоящее время в этом кластере генов мутации, ассоциированные с наследственными заболеваниями, найдены только в гене
- 43. Тип заболевания IA является наиболее тяжелой аутосомно-рецессивной формой карликовости, при которой гормон роста полностью отсутствует. Причиной
- 44. В большинстве случаев задержка роста диагностируется уже в первом полугодии жизни. Часто у больных детей развивается
- 45. Экзогенный гормон роста при данном типе заболевания неэффективен. Более того, у больных в ответ на введение
- 46. При варианте недостаточности IВ содержание гормона роста в крови снижено, но все же определяется. Карликовость менее
- 47. При некоторых формах карликовости, сходных по своим клиническим проявлениям с вариантом IВ, у больных найдены мутации
- 48. Тип II семейной изолированной недостаточности гормона роста наследуется по доминантному типу и протекает также как тип
- 49. При аутосомно-доминантном синдроме Коварского уровень иммунореактивных форм гормона роста сохраняется в пределах нормы или даже выше,
- 50. При этом больные хорошо отвечают на терапию препаратами соматотропного гормона. Оказалось, что мутантный гормон роста, хотя
- 51. Мутации в гене рецептора гормона роста – GHR – также приводят к карликовости, известной как синдром
- 52. При синдроме Ларона наблюдается выраженная задержка роста, которая может быть очевидна уже при рождении. Кроме того,
- 53. Болезнь наследуется по аутосомно-рецессивному типу и чаще всего обусловлена мутациями с преждевременной терминацией трансляции. Среди них
- 54. Инактивирующие мутации в генах инсулиноподобного фактора роста 1 (IGF1) и его рецептора (IGF1R) приводят к двум
- 55. У таких больных наблюдается нормальный уровень гормона роста и его рецептора в сочетании с высоким уровнем
- 56. Пангипопитуитарная карликовость, или низкий рост в сочетании с дефицитом гормона роста, гонадотропинов, адренокортикотропного и тиреотропного гормонов,
- 57. Наряду с этим, описаны моногенные формы пангипопитуитаризма, обусловленные, в частности, нарушением регуляции синтеза и секреции гормонов
- 58. Мутации в этих генах приводят к комбинированной недостаточности гипофизарных гормонов (КНГГ), при которой у больных наблюдается
- 59. Так, полное отсутствие гормона роста и пролактина в сочетании с частичной недостаточностью тиреотропного гормона может быть
- 60. У больных с раннего детского возраста наблюдается грубая задержка роста, у некоторых развивается тяжелая умственная отсталость.
- 61. Другой аутосомно-рецессивный тип пангипопитуитарной карликовости, сочетающийся с гипогонадизмом, является результатом мутаций в гене PROP1. У таких
- 62. Причем подобная гормональная недостаточность у разных больных может появляться в разном возрасте При тяжелой гипоплазии гипофиза
- 63. Однако в большинстве случаев болезнь проявляется карликовостью, сходной с синдромом Ларона, в сочетании с выраженной задержкой
- 64. Мутации в гене LHX3 приводят к дефициту гормона роста и гонадотропина. Клинически болезнь проявляется в форме
- 65. Остальные типы пангипопитуитарной карликовости наследуются по аутосомно-доминантному типу. Мутации в гене LHX4 приводят к недостаточности соматотропина
- 66. Недостаточность гормонов гипофиза в сочетании с пороками развития ЦНС характерна для одного из наиболее распространенных типов
- 67. Наследственные болезни тиреоидного обмена
- 68. Одной из причин снижения функции щитовидной железы является врожденный гипотиреоз. Болезнь может развиваться еще во внутриутробном
- 69. При этом нарушается обмен мукополисахаридов и в тканях накапливается большое количество креатинина и муцинозного вещества, приводящих
- 70. Однако при раннем назначении больным гормонов щитовидной железы, в частности L-тироксина, можно предотвратить развитие инвалидизирующей симптоматики
- 71. Тяжелая форма врожденного гипотиреоза выявляется сразу после рождения ребенка из-за присутствия микседемы в сочетании с брадикардией,
- 72. Для больных характерны большая масса тела, увеличение языка, сухость, шелушение и бледность кожных покровов, холодных на
- 74. Наиболее яркая картина врожденного гипотиреоза проявляется к 4-6 месяцам жизни, особенно при естественном вскармливании. Дети начинают
- 75. В 85% случаев причиной наследственного врожденного гипотиреоза является агенезия, гипоплазия или чаще эктопическая локализация щитовидной железы.
- 76. Аутосомно-доминантные типы врожденного незобного гипотиреоза генетически гетерогенны. Высокий уровень тиреотропного гормона и снижение содержания тиреоидных гормонов
- 77. Мальформации щитовидной железы характерны для типов заболевания, обусловленных мутациями в генах транскрипционных факторов – PAX8 и
- 78. Продуктом гена CSX является кардиоспецифический транскрипционный фактор NKX2-5. Наследственные нарушения в его работе чаще всего обнаруживаются
- 79. Другие наследственные болезни тиреоидного обмена могут быть обусловлены нарушением органификации, транспорта или рециркуляции йода, а также
- 80. Наследственные формы ожирения
- 81. При ожирении наблюдается патологическое увеличение массы тела за счет жировой ткани. Этому способствует положительный энергетический баланс
- 82. Важная роль в поддержании энергетического равновесия принадлежит гормонам. Ожирение может развиваться при уменьшении затрат энергии, повышении
- 83. Поэтому ожирение может быть результатом как наследственных нарушений энергетического метаболизма, так и неправильного образа жизни, касающегося,
- 84. Предполагается, что изменчивость массы жира у человека на 30-50% обусловлена генетическими факторами. В большинстве случаев у
- 85. Гены-кандидаты, ассоциированные с ожирением, во многих случаях участвуют в контроле сигнального пути, ответственного за регуляцию количества
- 86. Действие лептина противоположно действию «гормона голода» - грелина. Количество лептина пропорционально объему жировой ткани. Из жировой
- 87. При этом активизируется метаболическая цепь, которая заканчивается выработкой меланокортина, снижающего потребление человеком пищи. Генетические нарушения любого
- 88. Мутации в гене лептина (LEP) и его рецептора (LEPR) приводят к редким аутосомно-рецессивным формам ожирения. Примерно
- 89. Этот прогормон является предшественником, по крайней мере, шести гормонов, включая АКТГ, липотропин, мелано-стимулирующие гормоны (альфа- и
- 90. У больных, наряду с ожирением, которое наблюдается уже в течение первых месяцев жизни, как правило, выявляется
- 91. К сходной форме ожирения приводят рецессивные мутации в гене прогормоновой конвертазы 1, участвующей в процессинге АКТГ
- 92. В этом случае у больных развивается гиперпроинсулинемия, так как эта конвертаза участвует в биогенезе инсулина, переводя
- 93. Однако наиболее частым является аутосомно-доминантный тип ожирения, обусловленный мутациями в гене рецептора 4 меланокортина – MC4R
- 94. Избыточная масса тела является одним из ведущих клинических проявлений многих наследственных синдромов – Прадера-Вилли, Альстрема, Барде-Бидля
- 95. Синдром Прадера-Вилли относится к болезням геномного импринтинга – у больных инактивированы локализованные в области 15q11-13 гены
- 96. После 6-месячного возраста развивается полифагия, ожирение. В пубертатном периоде отмечается проявление гипогонадотропного гипогонадизма, снижение когнитивных функций
- 98. При синдроме Барде-Бидля, ожирение может сочетаться с деградацией сетчатки глаз, поликистозом почек, гипогонадизмом, полидактилией и задержкой
- 99. Синдром Барде-Бидля– это гетерогенная группа аутосомно-рецессивных заболеваний. В настоящее время описаны 19 генетических типов этого синдрома,
- 100. Для многих генетических типов синдрома Барде-Бидля характерно «трехаллельное наследование» – присутствие гомозиготной или компаунд-гетерозиготной мутации в
- 101. При синдроме Альстрема ожирение с гиперинсулинемией сочетается с пигментной дегенерацией сетчатки, прогрессирующей нейросенсорной тугоухостью, дилатационной кардиомиопатией,
- 102. У больных с аутосомно-рецессивным синдромом Коэна при рождении наблюдается низкая масса тела, мышечная гипотония. В дальнейшем
- 103. Отмечаются характерные лицевые особенности – антимонголоидный разрез глаз, высокая спинка носа, постоянно открытый рот косоглазие. Болезнь
- 104. Сахарный диабет 1 и 2 типов
- 105. Сахарный диабет — это частое хроническое заболевание, которым страдает до 12% населения в странах Европы, Северной
- 106. При ИЗСД развивается абсолютная недостаточность инсулина, а особенностью патогенеза ИНСД является относительная недостаточность инсулина или инсулинорезистентность
- 107. ИЗСД отличает разнообразие этиологии и патогенеза, при этом в основе развития сахарного диабета 1 типа лежит
- 108. Характерными проявлениями заболевания являются жажда, полиурия, потеря массы тела, нарастающая общая слабость. Нередко ИЗСД манифестирует внезапно
- 109. Конкордантность среди монозиготных близнецов варьирует от 30% до 50%. Риск развития заболевания у брата или сестры
- 110. Значительная часть наследственной предрасположенности к ИЗСД формируется за счет присутствия специфических полиморфных аллелей в HLA-локусе. Чувствительность
- 111. Наиболее значимыми предрасполагающими аллелями являются DR3, DR4 и DQ-beta . При наличии соответствующих гаплотипов риск для
- 112. Другими значимыми генетическими факторами риска ИЗСД являются полиморфные аллели генов PTPN2, C12ORF30, ERBB3, KIAA0350
- 113. Сахарный диабет 2 типа обычно развивается в возрасте 40-60 лет. Вместе с тем, известны случаи развития
- 114. Сопутствующими этиотропными факторами ИНСД являются ожирение и полифагия, обуславливающими повышенную потребность в инсулине и, как следствие,
- 115. Это приводит к инсулиновой недостаточности, нарушению толерантности к глюкозе и развитию инсулинорезистентности тканей. Часто нарушение толерантности
- 116. В этиологии ИНСД генетические факторы могут определять секреторную недостаточность бета-клеток или резистентность рецепторов инсулина. Это гетерогенная
- 117. Показано, что в основе патогенеза подобных состояний лежат мутации в мтДНК. В некоторых семьях с материнским
- 118. В других семьях причиной наследуемого по материнской линии синдромального ИНСД являются точковые мутации в мтДНК. Во
- 119. MODY-диабет наследуется по аутосомно-доминантному типу. Он составляет 2-5% всех случаев ИНСД. Эта генетически гетерогенная патология связана
- 120. В настоящее время идентифицировано 11 генетических типов MODY-диабета. Из них наиболее частым является MODY2, обусловленный присутствием
- 121. Этот тип заболевания часто обнаруживается у детей с мягкой гипергликемией, а также у женщин с диабетом
- 122. Часто MODY-диабет может быть связан с нарушением транскрипционного контроля, обусловленного мутациями в генах гепатоцитарных ядерных факторов
- 123. При других генетических типах заболевания мутации найдены в генах панкреатического липолитического фермента (CEL); инсулина (INS); нерецепторной
- 124. Мутации в гене инсулинового рецептора (INSR) приводят к целой серии аллельных заболеваний, клинические проявления которых весьма
- 125. У больных могут наблюдаться как нарушение толерантности к глюкозе, так и типичная симптоматика ИНСД. В ряде
- 126. Лепречаунизм может проявляться задержкой внутриутробного развития, низким ростом и маленьким весом при рождении, отсутствием подкожной жировой
- 127. Синдром Рабсона-Менденхолла отличается более мягким течением и большей продолжительностью жизни. При инсулинорезистентности типа А больные доживают
- 128. Такое разное проявление мутаций в одном и том же гене INSR зависит от характера повреждения инсулинового
- 129. При мягких формах инсулинорезистентности чаще всего находят миссенс-мутации, причем в некоторых случаях они присутствуют у больных
- 130. Генетические причины развития многофакторного ИНСД очень разнообразны. В 25% случаев обнаруживается ассоциация заболевания с аллелями гена
- 131. Продукт этого гена участвует в формировании жировой ткани и перекисном окислении липидов. Мутации в гене PPARG
- 132. С повышенными частотами у больных присутствуют полиморфные аллели в генах транскрипционного фактора (PAX4), адипонектина (ADIPOQ), рецептора
- 133. Найдена ассоциация сахарного диабета 2 типа с полиморфными аллелями генов калиевых каналов (KCNQ1 и KCNJ15), транскрипционного
- 135. Скачать презентацию
Слайд 2Наследственные болезни эндокринной системы обусловлены мутациями в генах гормонов,
их рецепторов, а
Наследственные болезни эндокринной системы обусловлены мутациями в генах гормонов, их рецепторов, а

Слайд 3Так, различные синдромы гиперальдостеронизма и гипоальдостеронизма связаны
с избыточной продукцией или дефицитом
Так, различные синдромы гиперальдостеронизма и гипоальдостеронизма связаны с избыточной продукцией или дефицитом

Слайд 4Наследственная недостаточность гормона роста ассоциирована с различными вариантами карликовости, или нанизма.
Нарушения
Наследственная недостаточность гормона роста ассоциирована с различными вариантами карликовости, или нанизма. Нарушения

Слайд 5Аутоиммунное поражение островкового аппарата
поджелудочной железы, а также инсулинорезистентность инсулинозависимых тканей приводят
Аутоиммунное поражение островкового аппарата поджелудочной железы, а также инсулинорезистентность инсулинозависимых тканей приводят

Слайд 6В этиологии большинства случаев сахарного диабета участвуют как средовые, так и генетические
В этиологии большинства случаев сахарного диабета участвуют как средовые, так и генетические

Слайд 7Наследственный
альдостеронизм
Наследственный
альдостеронизм

Слайд 8Нарушения водно-солевого обмена являются ведущими клиническими проявлениями различных наследственных типов альдостеронизма.
Аутосомно-рецессивный
Нарушения водно-солевого обмена являются ведущими клиническими проявлениями различных наследственных типов альдостеронизма. Аутосомно-рецессивный

Слайд 9Этот ген кодирует полипептид 2 субсемейства 11b митохондриального цитохрома P-450, или альдостеронсинтетазу
Этот ген кодирует полипептид 2 субсемейства 11b митохондриального цитохрома P-450, или альдостеронсинтетазу

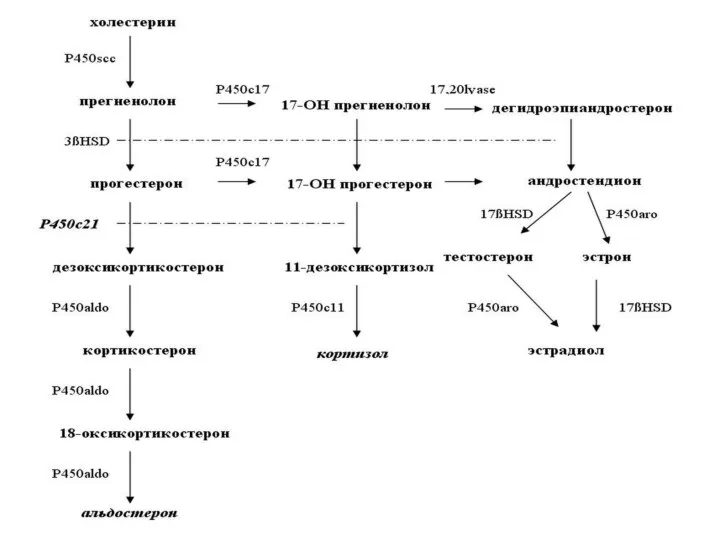
Слайд 11Как многие другие адреналовые цитохромы P-450, альдостеронсинтетаза
обладает множественной ферментативной активностью.
В
Как многие другие адреналовые цитохромы P-450, альдостеронсинтетаза обладает множественной ферментативной активностью. В

Слайд 12Мутации, затрагивающие разные активности фермента, приводят к двум аллельным вариантам заболевания с
Мутации, затрагивающие разные активности фермента, приводят к двум аллельным вариантам заболевания с

Слайд 13При биохимическом исследовании выявляются гипонатриемия, гиперкалиемия и ацидоз.
При этих заболеваниях, вызывающих
При биохимическом исследовании выявляются гипонатриемия, гиперкалиемия и ацидоз. При этих заболеваниях, вызывающих

Слайд 14Недостаточность КМОI и КМОII
ведет к накоплению прямых и отдаленных субстратов блокированных
Недостаточность КМОI и КМОII ведет к накоплению прямых и отдаленных субстратов блокированных

Слайд 15При первом варианте с недостаточностью КМОI болезнь проявляется в неонатальном периоде в
При первом варианте с недостаточностью КМОI болезнь проявляется в неонатальном периоде в

Слайд 16При втором варианте заболевания адреналовые кризы с выраженной потерей натрия и воды
При втором варианте заболевания адреналовые кризы с выраженной потерей натрия и воды

Слайд 17Наследственный гиперальдостеронизм является гетерогенной группой заболеваний.
Аутосомно-доминантный семейный гиперальдостеронизм I типа, или
Наследственный гиперальдостеронизм является гетерогенной группой заболеваний. Аутосомно-доминантный семейный гиперальдостеронизм I типа, или

Слайд 18Чаще этот тип гиперальдостеронизма рассматривается к одна из моногенных формам артериальной гипертензии
Чаще этот тип гиперальдостеронизма рассматривается к одна из моногенных формам артериальной гипертензии

Слайд 19Аутосомно-доминантный тип III заболевания обусловлен мутациями в гене KCNJ5, кодирующем субъединицу 5
Аутосомно-доминантный тип III заболевания обусловлен мутациями в гене KCNJ5, кодирующем субъединицу 5

Слайд 20Нарушения в работе других ионных каналов также могут приводить к различным формам
Нарушения в работе других ионных каналов также могут приводить к различным формам

Слайд 21Сопутствующими проявлениями заболевания являются артериальная гипертензия, легочная гипертензия и
врожденные пороки сердца
Сопутствующими проявлениями заболевания являются артериальная гипертензия, легочная гипертензия и
врожденные пороки сердца

Слайд 22Синдром Барттера является необычной формой
вторичного гиперальдостеронизма, при котором гипертрофия и гиперплазия
Синдром Барттера является необычной формой вторичного гиперальдостеронизма, при котором гипертрофия и гиперплазия

Слайд 23Болезнь развивается вследствие нарушения реабсорбции хлорида натрия в восходящем колене
петли Генле,
Болезнь развивается вследствие нарушения реабсорбции хлорида натрия в восходящем колене петли Генле,

Слайд 24Ведущими клиническими проявлениями заболевания являются задержка роста, гиперактивность ренин-ангиотензиновой системы, гипокалиемия, увеличение
Ведущими клиническими проявлениями заболевания являются задержка роста, гиперактивность ренин-ангиотензиновой системы, гипокалиемия, увеличение

Слайд 25Наследственные формы синдрома Барттера – это гетерогенная группа аутосомно-рецессивных заболеваний, вызванных нарушением
Наследственные формы синдрома Барттера – это гетерогенная группа аутосомно-рецессивных заболеваний, вызванных нарушением

Слайд 26Антенатальный тип 2 синдрома Барттера связан с мутациями в гене
АТФ-чувствительного
калиевого
Антенатальный тип 2 синдрома Барттера связан с мутациями в гене АТФ-чувствительного калиевого

Слайд 27 Причиной развития младенческого дигенного типа 4B синдрома Барттера, сочетающегося с нейросенсорной
Причиной развития младенческого дигенного типа 4B синдрома Барттера, сочетающегося с нейросенсорной

Слайд 28Псевдогипоальдостеронизм I типа обусловлен снижением чувствительности канальцевого эпителия к альдостерону и нарушением
Псевдогипоальдостеронизм I типа обусловлен снижением чувствительности канальцевого эпителия к альдостерону и нарушением

Слайд 29Типичными для этой патологии являются инфекционные поражения дыхательных путей. Компенсация водно-солевого обмена,
Типичными для этой патологии являются инфекционные поражения дыхательных путей. Компенсация водно-солевого обмена,

Слайд 30Наследственные формы заболевания генетически гетерогенны.
Аутосомно-доминантная форма псевдогипоальдостеронизма
типа IА с относительно
Наследственные формы заболевания генетически гетерогенны. Аутосомно-доминантная форма псевдогипоальдостеронизма типа IА с относительно

Слайд 31Причиной развития
аутосомно-рецессивного псевдогипоальдостеронизма типа IВ являются нарушения работы эпителиальных натриевых каналов,
Причиной развития аутосомно-рецессивного псевдогипоальдостеронизма типа IВ являются нарушения работы эпителиальных натриевых каналов,

Слайд 32Псевдогипоальдостеронизм II типа, известный как синдром Гордона, клинически характеризуется гиперкалиемией, гиперхлоремией, метаболическим
Псевдогипоальдостеронизм II типа, известный как синдром Гордона, клинически характеризуется гиперкалиемией, гиперхлоремией, метаболическим

Слайд 33Наследственные формы заболевания также генетически гетерогенны.
Два аутосомно-доминантных типа заболевания обусловлены мутациями
Наследственные формы заболевания также генетически гетерогенны. Два аутосомно-доминантных типа заболевания обусловлены мутациями

Слайд 34Другим регулятором этого котранспортера является транскрипционный фактор kelch3, кодируемый геном KLHL3.
Мутации
Другим регулятором этого котранспортера является транскрипционный фактор kelch3, кодируемый геном KLHL3. Мутации

Слайд 35Еще один аутосомно-доминантный тип заболевания обусловлен мутациями в гене куллина 3 –
Еще один аутосомно-доминантный тип заболевания обусловлен мутациями в гене куллина 3 –

Слайд 36Таким образом, все
генетические формы псевдогипоальдостеронизма
II типа
связаны с нарушением работы
Таким образом, все генетические формы псевдогипоальдостеронизма II типа связаны с нарушением работы

Слайд 37Гипофизарный нанизм
Гипофизарный нанизм

Слайд 38Рост скелета и мягких тканей организма индуцируется гормоном роста (соматотропином),
который синтезируется
Рост скелета и мягких тканей организма индуцируется гормоном роста (соматотропином), который синтезируется

Слайд 39После высвобождения из гипотоламуса биологически активный гормон роста связывается со своим трансмембранным
После высвобождения из гипотоламуса биологически активный гормон роста связывается со своим трансмембранным

Слайд 40Соматомедины являются членами инсулинового семейства полипептидных факторов роста и выполняют роль аутокринных
Соматомедины являются членами инсулинового семейства полипептидных факторов роста и выполняют роль аутокринных

Слайд 41Существует два гена гормона роста: нормальный — GH1, или GHN, и вариантный
Существует два гена гормона роста: нормальный — GH1, или GHN, и вариантный

Слайд 42В настоящее время в этом кластере генов мутации, ассоциированные с наследственными заболеваниями,
В настоящее время в этом кластере генов мутации, ассоциированные с наследственными заболеваниями,

Слайд 43Тип заболевания IA является наиболее тяжелой аутосомно-рецессивной формой карликовости, при которой гормон
Тип заболевания IA является наиболее тяжелой аутосомно-рецессивной формой карликовости, при которой гормон

Слайд 44В большинстве случаев задержка роста диагностируется уже в первом полугодии жизни. Часто
В большинстве случаев задержка роста диагностируется уже в первом полугодии жизни. Часто

Слайд 45Экзогенный гормон роста при данном типе заболевания неэффективен.
Более того, у больных
Экзогенный гормон роста при данном типе заболевания неэффективен. Более того, у больных

Слайд 46При варианте недостаточности IВ содержание гормона роста в крови снижено, но все
При варианте недостаточности IВ содержание гормона роста в крови снижено, но все

Слайд 47При некоторых формах карликовости, сходных по своим клиническим проявлениям с вариантом IВ,
При некоторых формах карликовости, сходных по своим клиническим проявлениям с вариантом IВ,

Слайд 48Тип II семейной изолированной недостаточности гормона роста наследуется по доминантному типу и
Тип II семейной изолированной недостаточности гормона роста наследуется по доминантному типу и

Слайд 49При аутосомно-доминантном синдроме Коварского уровень иммунореактивных форм гормона роста сохраняется в пределах
При аутосомно-доминантном синдроме Коварского уровень иммунореактивных форм гормона роста сохраняется в пределах

Слайд 50При этом больные хорошо отвечают на терапию препаратами соматотропного гормона. Оказалось, что
При этом больные хорошо отвечают на терапию препаратами соматотропного гормона. Оказалось, что

Слайд 51Мутации в гене
рецептора гормона роста – GHR – также приводят к
Мутации в гене рецептора гормона роста – GHR – также приводят к

Слайд 52При синдроме Ларона наблюдается выраженная задержка роста, которая может быть очевидна уже
При синдроме Ларона наблюдается выраженная задержка роста, которая может быть очевидна уже

Слайд 53Болезнь наследуется по аутосомно-рецессивному типу и чаще всего обусловлена мутациями с преждевременной
Болезнь наследуется по аутосомно-рецессивному типу и чаще всего обусловлена мутациями с преждевременной

Слайд 54Инактивирующие мутации в генах инсулиноподобного фактора роста 1 (IGF1) и его рецептора
Инактивирующие мутации в генах инсулиноподобного фактора роста 1 (IGF1) и его рецептора

Слайд 55У таких больных наблюдается нормальный уровень гормона роста и его рецептора в
У таких больных наблюдается нормальный уровень гормона роста и его рецептора в

Слайд 56Пангипопитуитарная карликовость,
или низкий рост в сочетании с дефицитом гормона роста, гонадотропинов,
Пангипопитуитарная карликовость, или низкий рост в сочетании с дефицитом гормона роста, гонадотропинов,

Слайд 57Наряду с этим, описаны моногенные формы пангипопитуитаризма, обусловленные, в частности, нарушением регуляции
Наряду с этим, описаны моногенные формы пангипопитуитаризма, обусловленные, в частности, нарушением регуляции

Слайд 58Мутации в этих генах приводят к комбинированной недостаточности гипофизарных гормонов (КНГГ), при
Мутации в этих генах приводят к комбинированной недостаточности гипофизарных гормонов (КНГГ), при

Слайд 59Так, полное отсутствие
гормона роста и пролактина
в сочетании с частичной недостаточностью
Так, полное отсутствие гормона роста и пролактина в сочетании с частичной недостаточностью

Слайд 60У больных с раннего детского возраста наблюдается грубая задержка роста,
у некоторых
У больных с раннего детского возраста наблюдается грубая задержка роста, у некоторых

Слайд 61Другой аутосомно-рецессивный тип пангипопитуитарной карликовости, сочетающийся с гипогонадизмом, является результатом мутаций в
Другой аутосомно-рецессивный тип пангипопитуитарной карликовости, сочетающийся с гипогонадизмом, является результатом мутаций в

Слайд 62Причем подобная гормональная недостаточность у разных больных может появляться в разном возрасте
Причем подобная гормональная недостаточность у разных больных может появляться в разном возрасте

Слайд 63Однако в большинстве случаев болезнь проявляется карликовостью, сходной с синдромом Ларона, в
Однако в большинстве случаев болезнь проявляется карликовостью, сходной с синдромом Ларона, в

Слайд 64Мутации в гене LHX3 приводят к дефициту гормона роста и гонадотропина.
Клинически
Мутации в гене LHX3 приводят к дефициту гормона роста и гонадотропина. Клинически

Слайд 65Остальные типы пангипопитуитарной карликовости наследуются по аутосомно-доминантному типу. Мутации в гене LHX4
Остальные типы пангипопитуитарной карликовости наследуются по аутосомно-доминантному типу. Мутации в гене LHX4

Слайд 66Недостаточность гормонов гипофиза в сочетании с пороками развития ЦНС характерна для одного
Недостаточность гормонов гипофиза в сочетании с пороками развития ЦНС характерна для одного

Слайд 67Наследственные болезни тиреоидного обмена
Наследственные болезни тиреоидного обмена

Слайд 68Одной из причин снижения функции щитовидной железы является врожденный гипотиреоз. Болезнь может
Одной из причин снижения функции щитовидной железы является врожденный гипотиреоз. Болезнь может

Слайд 69При этом нарушается обмен мукополисахаридов и в тканях накапливается большое количество креатинина
При этом нарушается обмен мукополисахаридов и в тканях накапливается большое количество креатинина

Слайд 70Однако при раннем назначении больным гормонов щитовидной железы, в частности L-тироксина, можно
Однако при раннем назначении больным гормонов щитовидной железы, в частности L-тироксина, можно

Слайд 71Тяжелая форма врожденного гипотиреоза выявляется сразу после рождения ребенка из-за присутствия микседемы
Тяжелая форма врожденного гипотиреоза выявляется сразу после рождения ребенка из-за присутствия микседемы

Слайд 72Для больных характерны большая масса тела, увеличение языка, сухость, шелушение и бледность
Для больных характерны большая масса тела, увеличение языка, сухость, шелушение и бледность

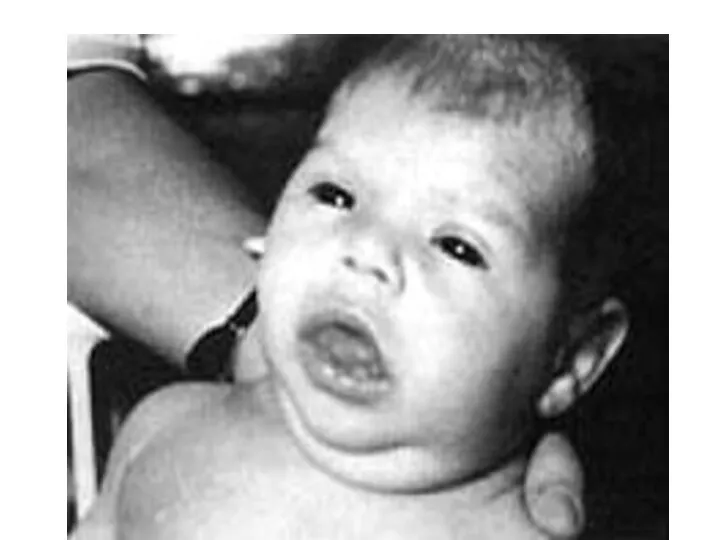
Слайд 74Наиболее яркая картина врожденного гипотиреоза проявляется к 4-6 месяцам жизни, особенно при
Наиболее яркая картина врожденного гипотиреоза проявляется к 4-6 месяцам жизни, особенно при

Слайд 75В 85% случаев причиной наследственного врожденного гипотиреоза является агенезия, гипоплазия или чаще
В 85% случаев причиной наследственного врожденного гипотиреоза является агенезия, гипоплазия или чаще

Слайд 76Аутосомно-доминантные типы врожденного незобного гипотиреоза генетически гетерогенны.
Высокий уровень тиреотропного гормона и
Аутосомно-доминантные типы врожденного незобного гипотиреоза генетически гетерогенны. Высокий уровень тиреотропного гормона и

Слайд 77Мальформации щитовидной железы характерны для типов заболевания, обусловленных мутациями в генах транскрипционных
Мальформации щитовидной железы характерны для типов заболевания, обусловленных мутациями в генах транскрипционных

Слайд 78Продуктом гена CSX является кардиоспецифический транскрипционный фактор NKX2-5. Наследственные нарушения в его
Продуктом гена CSX является кардиоспецифический транскрипционный фактор NKX2-5. Наследственные нарушения в его

Слайд 79Другие наследственные болезни тиреоидного обмена могут быть обусловлены нарушением органификации, транспорта или
Другие наследственные болезни тиреоидного обмена могут быть обусловлены нарушением органификации, транспорта или

Слайд 80Наследственные формы ожирения
Наследственные формы ожирения

Слайд 81При ожирении наблюдается патологическое увеличение массы тела за счет жировой ткани.
Этому
При ожирении наблюдается патологическое увеличение массы тела за счет жировой ткани. Этому

Слайд 82Важная роль в поддержании энергетического равновесия принадлежит гормонам.
Ожирение может развиваться при
Важная роль в поддержании энергетического равновесия принадлежит гормонам. Ожирение может развиваться при

Слайд 83Поэтому ожирение может быть результатом как наследственных нарушений энергетического метаболизма, так и
Поэтому ожирение может быть результатом как наследственных нарушений энергетического метаболизма, так и

Слайд 84Предполагается, что изменчивость массы жира у человека на 30-50% обусловлена генетическими факторами.
Предполагается, что изменчивость массы жира у человека на 30-50% обусловлена генетическими факторами.

Слайд 85Гены-кандидаты, ассоциированные с ожирением, во многих случаях участвуют в контроле сигнального пути,
Гены-кандидаты, ассоциированные с ожирением, во многих случаях участвуют в контроле сигнального пути,

Слайд 86Действие лептина противоположно действию «гормона голода» - грелина. Количество лептина пропорционально объему
Действие лептина противоположно действию «гормона голода» - грелина. Количество лептина пропорционально объему

Слайд 87При этом активизируется метаболическая цепь, которая заканчивается выработкой меланокортина, снижающего потребление человеком
При этом активизируется метаболическая цепь, которая заканчивается выработкой меланокортина, снижающего потребление человеком

Слайд 88Мутации в гене лептина (LEP) и его рецептора (LEPR) приводят к редким
Мутации в гене лептина (LEP) и его рецептора (LEPR) приводят к редким

Слайд 89Этот прогормон является предшественником, по крайней мере, шести гормонов, включая АКТГ, липотропин,
Этот прогормон является предшественником, по крайней мере, шести гормонов, включая АКТГ, липотропин,

Слайд 90У больных, наряду с ожирением, которое наблюдается уже в течение первых месяцев
У больных, наряду с ожирением, которое наблюдается уже в течение первых месяцев

Слайд 91К сходной форме ожирения приводят рецессивные мутации в гене
прогормоновой конвертазы 1,
К сходной форме ожирения приводят рецессивные мутации в гене прогормоновой конвертазы 1,

Слайд 92В этом случае у больных развивается гиперпроинсулинемия, так как эта конвертаза участвует
В этом случае у больных развивается гиперпроинсулинемия, так как эта конвертаза участвует

Слайд 93Однако наиболее частым является аутосомно-доминантный тип ожирения, обусловленный мутациями в гене рецептора
Однако наиболее частым является аутосомно-доминантный тип ожирения, обусловленный мутациями в гене рецептора

Слайд 94Избыточная масса тела является одним из ведущих клинических проявлений многих наследственных синдромов
Избыточная масса тела является одним из ведущих клинических проявлений многих наследственных синдромов

Слайд 95Синдром Прадера-Вилли относится к болезням геномного импринтинга –
у больных инактивированы локализованные
Синдром Прадера-Вилли относится к болезням геномного импринтинга – у больных инактивированы локализованные

Слайд 96После 6-месячного возраста развивается полифагия, ожирение.
В пубертатном периоде отмечается проявление гипогонадотропного
После 6-месячного возраста развивается полифагия, ожирение. В пубертатном периоде отмечается проявление гипогонадотропного


Слайд 98При синдроме Барде-Бидля,
ожирение может сочетаться с деградацией сетчатки глаз, поликистозом почек,
При синдроме Барде-Бидля, ожирение может сочетаться с деградацией сетчатки глаз, поликистозом почек,

Слайд 99Синдром Барде-Бидля–
это гетерогенная группа аутосомно-рецессивных заболеваний.
В настоящее время описаны 19
Синдром Барде-Бидля– это гетерогенная группа аутосомно-рецессивных заболеваний. В настоящее время описаны 19

Слайд 100Для многих генетических типов синдрома Барде-Бидля характерно «трехаллельное наследование» – присутствие гомозиготной
Для многих генетических типов синдрома Барде-Бидля характерно «трехаллельное наследование» – присутствие гомозиготной

Слайд 101При синдроме Альстрема ожирение с гиперинсулинемией сочетается с пигментной дегенерацией сетчатки, прогрессирующей
При синдроме Альстрема ожирение с гиперинсулинемией сочетается с пигментной дегенерацией сетчатки, прогрессирующей

Слайд 102У больных с аутосомно-рецессивным синдромом Коэна при рождении наблюдается низкая масса тела,
У больных с аутосомно-рецессивным синдромом Коэна при рождении наблюдается низкая масса тела,

Слайд 103Отмечаются характерные лицевые особенности – антимонголоидный разрез глаз, высокая спинка носа, постоянно
Отмечаются характерные лицевые особенности – антимонголоидный разрез глаз, высокая спинка носа, постоянно

Слайд 104Сахарный диабет 1 и 2 типов
Сахарный диабет 1 и 2 типов

Слайд 105Сахарный диабет — это частое хроническое заболевание, которым страдает до 12% населения
Сахарный диабет — это частое хроническое заболевание, которым страдает до 12% населения

Слайд 106При ИЗСД развивается абсолютная недостаточность инсулина, а особенностью патогенеза ИНСД является относительная
При ИЗСД развивается абсолютная недостаточность инсулина, а особенностью патогенеза ИНСД является относительная

Слайд 107ИЗСД отличает разнообразие этиологии и патогенеза, при этом в основе развития сахарного
ИЗСД отличает разнообразие этиологии и патогенеза, при этом в основе развития сахарного

Слайд 108Характерными проявлениями заболевания являются жажда, полиурия, потеря массы тела, нарастающая общая слабость.
Характерными проявлениями заболевания являются жажда, полиурия, потеря массы тела, нарастающая общая слабость.

Слайд 109Конкордантность среди монозиготных близнецов варьирует от 30% до 50%.
Риск развития заболевания
Конкордантность среди монозиготных близнецов варьирует от 30% до 50%. Риск развития заболевания

Слайд 110Значительная часть наследственной предрасположенности к ИЗСД формируется за счет присутствия специфических полиморфных
Значительная часть наследственной предрасположенности к ИЗСД формируется за счет присутствия специфических полиморфных

Слайд 111Наиболее значимыми предрасполагающими аллелями являются DR3, DR4 и DQ-beta . При наличии
Наиболее значимыми предрасполагающими аллелями являются DR3, DR4 и DQ-beta . При наличии

Слайд 112 Другими значимыми генетическими факторами риска ИЗСД являются полиморфные аллели генов
PTPN2,
Другими значимыми генетическими факторами риска ИЗСД являются полиморфные аллели генов PTPN2,

Слайд 113Сахарный диабет 2 типа обычно развивается в возрасте 40-60 лет. Вместе с
Сахарный диабет 2 типа обычно развивается в возрасте 40-60 лет. Вместе с

Слайд 114Сопутствующими этиотропными факторами ИНСД являются ожирение и полифагия, обуславливающими повышенную потребность в
Сопутствующими этиотропными факторами ИНСД являются ожирение и полифагия, обуславливающими повышенную потребность в

Слайд 115Это приводит к инсулиновой недостаточности, нарушению толерантности к глюкозе и развитию инсулинорезистентности
Это приводит к инсулиновой недостаточности, нарушению толерантности к глюкозе и развитию инсулинорезистентности

Слайд 116В этиологии ИНСД генетические факторы могут определять секреторную недостаточность бета-клеток или резистентность
В этиологии ИНСД генетические факторы могут определять секреторную недостаточность бета-клеток или резистентность

Слайд 117 Показано, что в основе патогенеза подобных состояний лежат
мутации в мтДНК.
Показано, что в основе патогенеза подобных состояний лежат мутации в мтДНК.

Слайд 118В других семьях причиной наследуемого по материнской линии синдромального ИНСД являются точковые
В других семьях причиной наследуемого по материнской линии синдромального ИНСД являются точковые

Слайд 119MODY-диабет
наследуется по аутосомно-доминантному типу.
Он составляет 2-5% всех случаев ИНСД.
Эта
MODY-диабет наследуется по аутосомно-доминантному типу. Он составляет 2-5% всех случаев ИНСД. Эта

Слайд 120В настоящее время идентифицировано 11 генетических типов
MODY-диабета.
Из них наиболее частым
В настоящее время идентифицировано 11 генетических типов MODY-диабета. Из них наиболее частым

Слайд 121Этот тип заболевания часто обнаруживается у детей с мягкой гипергликемией, а также
Этот тип заболевания часто обнаруживается у детей с мягкой гипергликемией, а также

Слайд 122Часто MODY-диабет может быть связан с нарушением транскрипционного контроля, обусловленного мутациями в
Часто MODY-диабет может быть связан с нарушением транскрипционного контроля, обусловленного мутациями в

Слайд 123При других генетических типах заболевания мутации найдены в генах панкреатического липолитического фермента
При других генетических типах заболевания мутации найдены в генах панкреатического липолитического фермента

Слайд 124Мутации в гене инсулинового рецептора (INSR) приводят к целой серии аллельных заболеваний,
Мутации в гене инсулинового рецептора (INSR) приводят к целой серии аллельных заболеваний,

Слайд 125У больных могут наблюдаться как нарушение толерантности к глюкозе, так и типичная
У больных могут наблюдаться как нарушение толерантности к глюкозе, так и типичная

Слайд 126Лепречаунизм может проявляться задержкой внутриутробного развития, низким ростом и маленьким весом при
Лепречаунизм может проявляться задержкой внутриутробного развития, низким ростом и маленьким весом при

Слайд 127Синдром Рабсона-Менденхолла отличается более мягким течением и большей продолжительностью жизни. При инсулинорезистентности
Синдром Рабсона-Менденхолла отличается более мягким течением и большей продолжительностью жизни. При инсулинорезистентности

Слайд 128Такое разное проявление мутаций в одном и том же гене INSR зависит
Такое разное проявление мутаций в одном и том же гене INSR зависит

Слайд 129При мягких формах инсулинорезистентности чаще всего находят миссенс-мутации, причем в некоторых случаях
При мягких формах инсулинорезистентности чаще всего находят миссенс-мутации, причем в некоторых случаях

Слайд 130Генетические причины развития многофакторного ИНСД очень разнообразны.
В 25% случаев обнаруживается ассоциация
Генетические причины развития многофакторного ИНСД очень разнообразны. В 25% случаев обнаруживается ассоциация

Слайд 131Продукт этого гена участвует в формировании жировой ткани и перекисном окислении липидов.
Продукт этого гена участвует в формировании жировой ткани и перекисном окислении липидов.

Слайд 132С повышенными частотами у больных присутствуют полиморфные аллели в генах транскрипционного фактора
С повышенными частотами у больных присутствуют полиморфные аллели в генах транскрипционного фактора

Слайд 133Найдена ассоциация сахарного диабета 2 типа с полиморфными аллелями генов калиевых каналов
Найдена ассоциация сахарного диабета 2 типа с полиморфными аллелями генов калиевых каналов

 Виды портрета человека (6 класс)
Виды портрета человека (6 класс) Шерстяная ткань
Шерстяная ткань Александр Васильевич Колчак
Александр Васильевич Колчак ЦЕНЫ НИЖЕ!
ЦЕНЫ НИЖЕ! ВКР: Социальное страхование в России: основные направления и пути совершенствования
ВКР: Социальное страхование в России: основные направления и пути совершенствования STG
STG Псалом 117, мессианский
Псалом 117, мессианский Рисуем животных вместе
Рисуем животных вместе Спортивные комплексы района
Спортивные комплексы района Изготовление декоративного цветка
Изготовление декоративного цветка Роль государства в развитии Отечественного спорта в период 1990-х – 2000-х гг
Роль государства в развитии Отечественного спорта в период 1990-х – 2000-х гг Четыре живописца
Четыре живописца Презентация на тему Биологическое разнообразие Москвы, флора и фауна
Презентация на тему Биологическое разнообразие Москвы, флора и фауна Ну-ка, проверь-ка, дружок, Ты готов начать урок? Всё ль на месте, всё ль в порядке, Ручка, книжка и тетрадка? Все ли правильно сидят? Вс
Ну-ка, проверь-ка, дружок, Ты готов начать урок? Всё ль на месте, всё ль в порядке, Ручка, книжка и тетрадка? Все ли правильно сидят? Вс ВЫСШАЯ ШКОЛА МАРКЕТИНГА И РАЗВИТИЯ БИЗНЕСА Презентация пилотного исследования «Действия российских компаний направленные на уп
ВЫСШАЯ ШКОЛА МАРКЕТИНГА И РАЗВИТИЯ БИЗНЕСА Презентация пилотного исследования «Действия российских компаний направленные на уп Способы резки и раскроя металла
Способы резки и раскроя металла Структура бизнес-предложения
Структура бизнес-предложения Совершенствование учительского корпуса
Совершенствование учительского корпуса Презентация на тему Устное народное творчество 3 класс
Презентация на тему Устное народное творчество 3 класс b91c09cb9abc704a
b91c09cb9abc704a Консервативное и миниинвазивное лечение гемангиом печени
Консервативное и миниинвазивное лечение гемангиом печени Электросамокаты
Электросамокаты Презентация на тему:
Презентация на тему: Automation Cloud Infrastructure, или откуда в России появится свой Amazon
Automation Cloud Infrastructure, или откуда в России появится свой Amazon Сольфеджио для детей
Сольфеджио для детей Натюрморт с фруктами
Натюрморт с фруктами Результаты ЕГЭ и ГИА Поступление в вузы 2010-2011 учебный год
Результаты ЕГЭ и ГИА Поступление в вузы 2010-2011 учебный год Экологизация школьного курса окружающего мира
Экологизация школьного курса окружающего мира