- Презентация на тему Методы исследований генетики человека

Содержание
- 2. Генеалогический метод. Разработан в 1865 году Ф. Гальтоном. Задачи метода: Определение наследственного характера признака. Определение типа
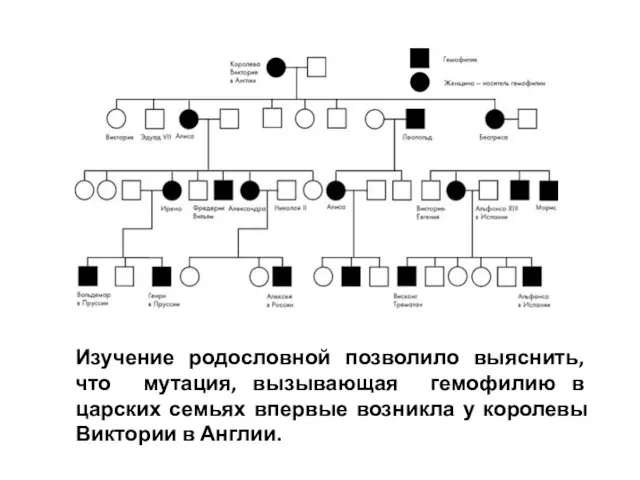
- 3. Изучение родословной позволило выяснить, что мутация, вызывающая гемофилию в царских семьях впервые возникла у королевы Виктории
- 4. Цитогенетический метод. Изучение структуры и числа хромосом.
- 5. Хромосомные заболевания. Синдром Дауна ( трисомия по 21 хромосоме)
- 6. Синдром Дауна возникает в результате генетической аномалии. Впервые признаки людей с синдромом Дауна описал в 1866
- 7. Синдром Патау (трисомия по 13 хромосоме) Трисомия 13 впервые была описана Томасом Бартолини в 1657 году,
- 8. ПОРОКИ РАЗВИТИЯ Нервная система: - отклонения психического и моторного развития; - микроцефалия; - голопрозэнцефалия (нарушение формирования
- 9. Синдром Эдварса(трисомия по 18 хромосоме) Синдром Эдвардса был назван в честь доктора Джона Эдварда, который в
- 10. ПОРОКИ РАЗВИТИЯ Низкий вес при рождении независимо от срока беременности; Характерные изменения головы: деформированный маленький череп,
- 11. Трисомии по половым хромосомам.
- 12. Синдром Клайнфельтера — это генетическое заболевание у лиц мужского пола, в основе которого лежит генетически обусловленный
- 13. Близнецовый метод. В 1876 году Ф. Гальтон предложил использовать метод анализа близнецов для разграничения роли наследственности
- 14. Виды однояйцовых близнецов.
- 19. Сиамские близнецы. Сиа́мские близнецы́ — это однояйцовые близнецы, которые не полностью разделились в эмбриональном периоде развития
- 20. Возможно, наиболее знаменитой парой близнецов были китайцы Чанг и Энг Банкеры (1811—1874), родившиеся в Сиаме (современный
- 23. Медико-генетическое консультирование. Цель: Предупреждение рождения ребенка с тяжелыми наследственными заболеваниями. Работают во всех крупных городах России.
- 25. Пренатальная диагностика. Она использует и ультразвуковую диагностику (УЗИ), и оперативную технику и лабораторные методы(цитогенетические, биохимические, молекулярно-генетические).
- 28. Скачать презентацию
Слайд 2Генеалогический метод.
Разработан в 1865 году Ф. Гальтоном.
Задачи метода:
Определение наследственного характера признака.
Определение
Генеалогический метод.
Разработан в 1865 году Ф. Гальтоном.
Задачи метода:
Определение наследственного характера признака.
Определение

Слайд 3Изучение родословной позволило выяснить, что мутация, вызывающая гемофилию в царских семьях впервые
Изучение родословной позволило выяснить, что мутация, вызывающая гемофилию в царских семьях впервые
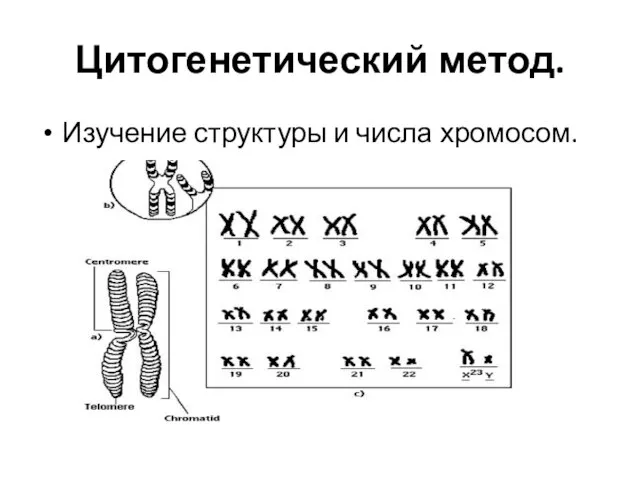
Слайд 4Цитогенетический метод.
Изучение структуры и числа хромосом.
Цитогенетический метод.
Изучение структуры и числа хромосом.
Слайд 5Хромосомные заболевания.
Синдром Дауна ( трисомия по 21 хромосоме)
Хромосомные заболевания.
Синдром Дауна ( трисомия по 21 хромосоме)

Слайд 6Синдром Дауна возникает в результате генетической аномалии. Впервые признаки людей с синдромом
Синдром Дауна возникает в результате генетической аномалии. Впервые признаки людей с синдромом

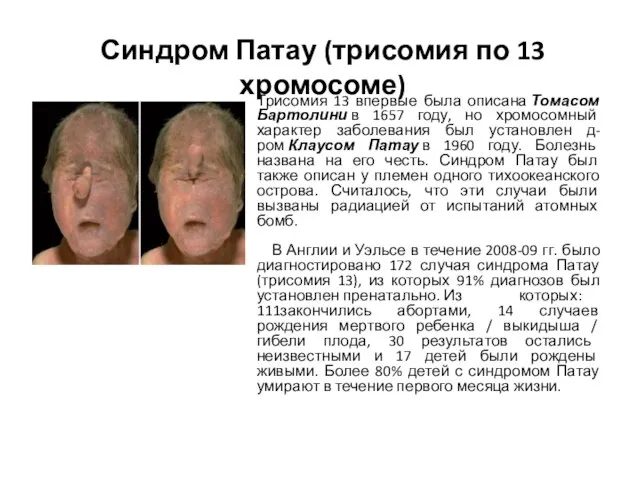
Слайд 7Синдром Патау (трисомия по 13 хромосоме)
Трисомия 13 впервые была описана Томасом Бартолини в 1657
Синдром Патау (трисомия по 13 хромосоме)
Трисомия 13 впервые была описана Томасом Бартолини в 1657
Слайд 8
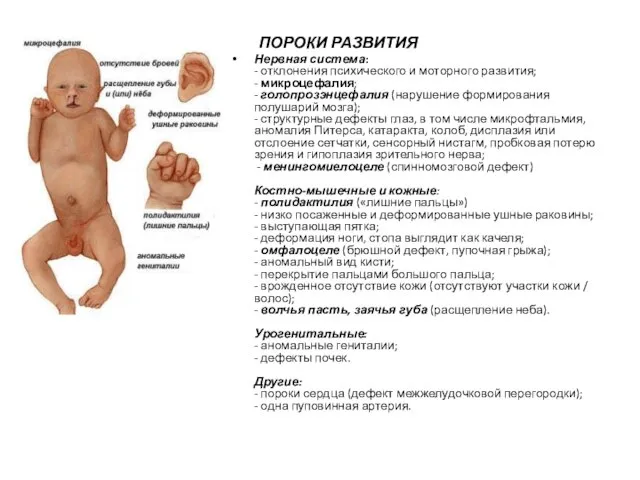
ПОРОКИ РАЗВИТИЯ
Нервная система:
- отклонения психического и моторного развития;
- микроцефалия;
- голопрозэнцефалия (нарушение формирования полушарий мозга);
-
ПОРОКИ РАЗВИТИЯ
Нервная система:
- отклонения психического и моторного развития;
- микроцефалия;
- голопрозэнцефалия (нарушение формирования полушарий мозга);
-
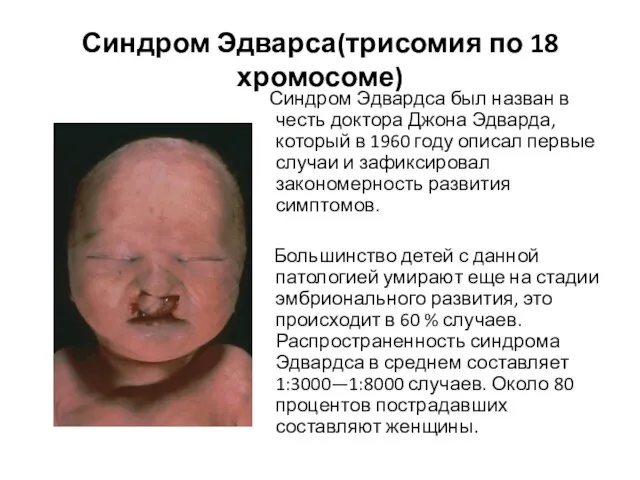
Слайд 9Синдром Эдварса(трисомия по 18 хромосоме)
Синдром Эдвардса был назван в честь доктора
Синдром Эдварса(трисомия по 18 хромосоме)
Синдром Эдвардса был назван в честь доктора
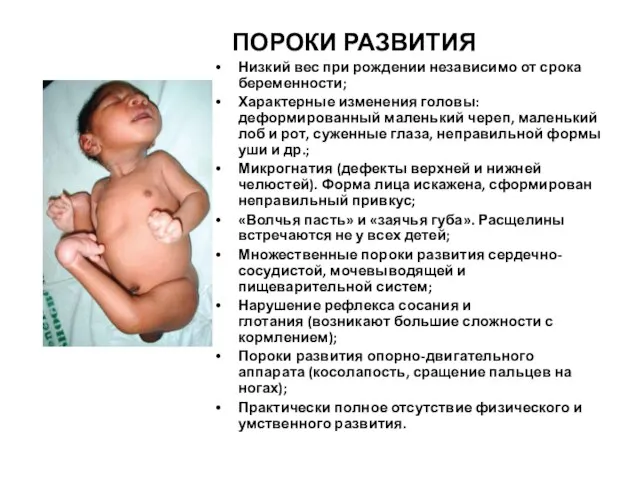
Слайд 10 ПОРОКИ РАЗВИТИЯ
Низкий вес при рождении независимо от срока беременности;
Характерные изменения головы:
ПОРОКИ РАЗВИТИЯ
Низкий вес при рождении независимо от срока беременности;
Характерные изменения головы:
Слайд 11 Трисомии по половым хромосомам.
Трисомии по половым хромосомам.

Слайд 12Синдром Клайнфельтера — это генетическое заболевание у лиц мужского пола, в основе
Синдром Клайнфельтера — это генетическое заболевание у лиц мужского пола, в основе

Слайд 13Близнецовый метод.
В 1876 году Ф. Гальтон предложил использовать метод анализа близнецов для
Близнецовый метод.
В 1876 году Ф. Гальтон предложил использовать метод анализа близнецов для

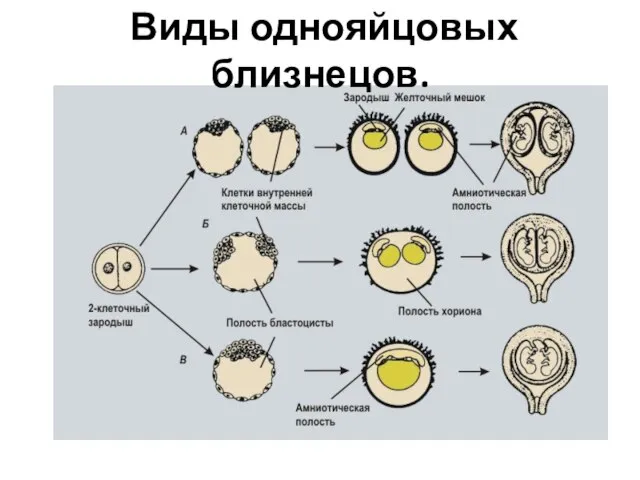
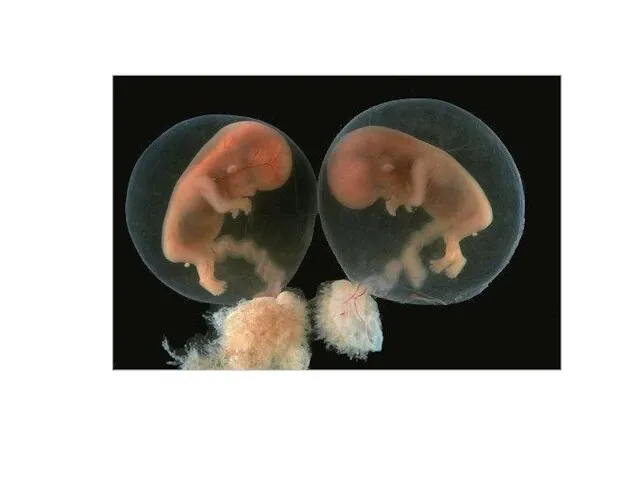
Слайд 14 Виды однояйцовых близнецов.
Виды однояйцовых близнецов.



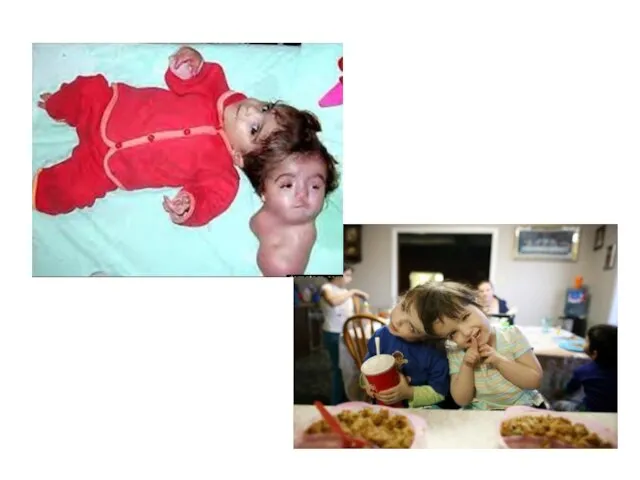
Слайд 19 Сиамские близнецы.
Сиа́мские близнецы́ — это однояйцовые близнецы, которые не полностью разделились в
Сиамские близнецы.
Сиа́мские близнецы́ — это однояйцовые близнецы, которые не полностью разделились в

Слайд 20Возможно, наиболее знаменитой парой близнецов были китайцы Чанг и Энг Банкеры (1811—1874),

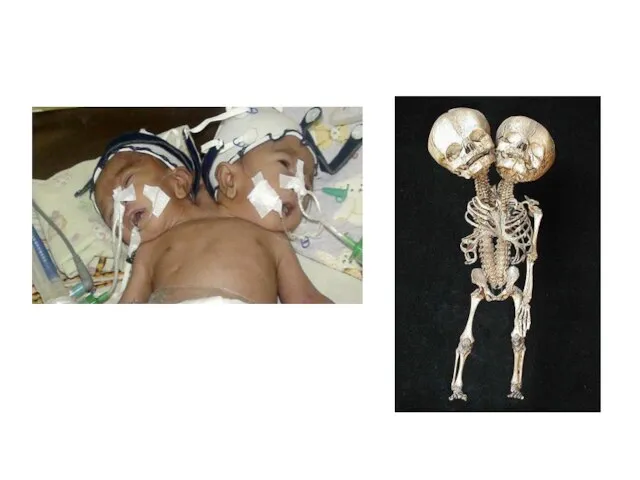
Слайд 23Медико-генетическое консультирование.
Цель: Предупреждение рождения ребенка с тяжелыми наследственными заболеваниями.
Работают во всех
Медико-генетическое консультирование.
Цель: Предупреждение рождения ребенка с тяжелыми наследственными заболеваниями.
Работают во всех


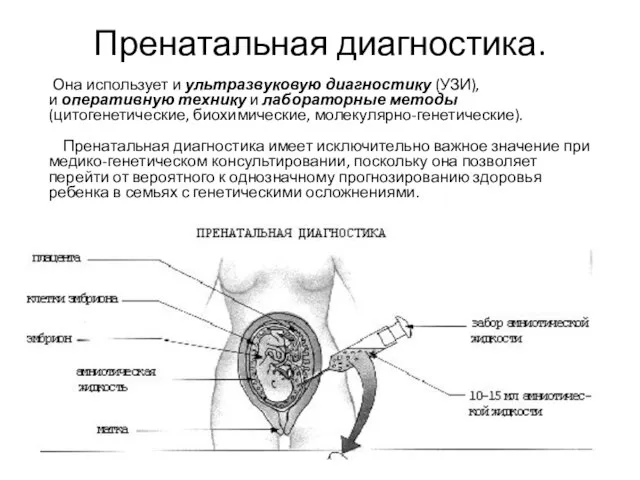
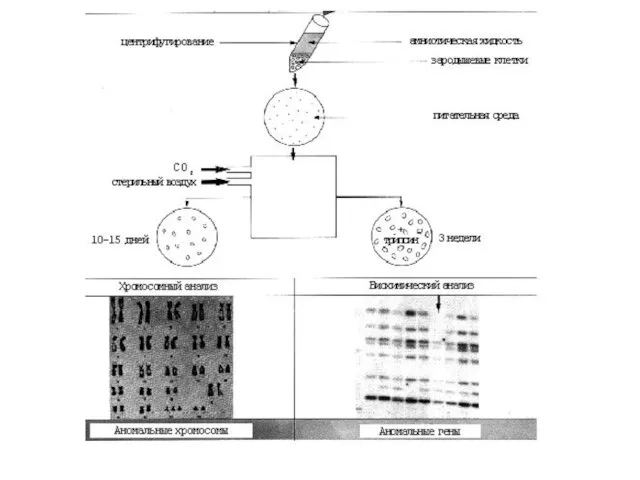
Слайд 25Пренатальная диагностика.
Она использует и ультразвуковую диагностику (УЗИ), и оперативную технику и лабораторные методы(цитогенетические, биохимические, молекулярно-генетические).
Пренатальная
Пренатальная диагностика.
Она использует и ультразвуковую диагностику (УЗИ), и оперативную технику и лабораторные методы(цитогенетические, биохимические, молекулярно-генетические). Пренатальная
 Презентация на тему Полоролевое развитие детей старшего дошкольного возраста
Презентация на тему Полоролевое развитие детей старшего дошкольного возраста  Министерство юстиции и его органы на местах. Прокуратура Республики Беларусь. Тема 9-10
Министерство юстиции и его органы на местах. Прокуратура Республики Беларусь. Тема 9-10 Портфолио учащегося
Портфолио учащегося Сульфур
Сульфур Функциональные возможности дыхательной системы
Функциональные возможности дыхательной системы Храмы Кубани
Храмы Кубани Как выбрать CMS для сайта
Как выбрать CMS для сайта Лекция 4 Планирование деятельности
Лекция 4 Планирование деятельности Жизненный и творческий путь Ивана Александровича Гончарова 1812 – 1891 г.г
Жизненный и творческий путь Ивана Александровича Гончарова 1812 – 1891 г.г Жизненные формы в растительном мире
Жизненные формы в растительном мире ТЕХНИКО-ЭКОНОМИЧЕСКИЕ ВОПРОСЫ РЕГРЕССИОННОГО ФУНКЦИОНАЛЬНОГО И НАГРУЗОЧНОГО ТЕСИРОВАНИЯ ПРИ СОПРОВОЖДЕНИИ И ЭКСПЛУАТАЦИИ АВТОМ
ТЕХНИКО-ЭКОНОМИЧЕСКИЕ ВОПРОСЫ РЕГРЕССИОННОГО ФУНКЦИОНАЛЬНОГО И НАГРУЗОЧНОГО ТЕСИРОВАНИЯ ПРИ СОПРОВОЖДЕНИИ И ЭКСПЛУАТАЦИИ АВТОМ Internet Explorer для разработчика
Internet Explorer для разработчика Консерватизм
Консерватизм Профессия эколог
Профессия эколог БуСтройМаркет
БуСтройМаркет Сторителлинг Пишем с пользой!
Сторителлинг Пишем с пользой! Воздух. Свойства воздуха
Воздух. Свойства воздуха Правописание корней. Чередование гласных в корнях слов
Правописание корней. Чередование гласных в корнях слов ПРАВОВОЕ ГОСУДАРСТВО
ПРАВОВОЕ ГОСУДАРСТВО Живое кино
Живое кино Таможенник – представитель РФ
Таможенник – представитель РФ A guide to the Ukrainian cuisine
A guide to the Ukrainian cuisine Болезни на английском
Болезни на английском Образ матери в русской литературе
Образ матери в русской литературе Трудовой договор: понятие, содержание, порядок заключения и расторжения
Трудовой договор: понятие, содержание, порядок заключения и расторжения Сборный портфель – одна из форм учета учебных достижений первоклассников в условиях безотметочного обучения
Сборный портфель – одна из форм учета учебных достижений первоклассников в условиях безотметочного обучения Dept. Research Themes
Dept. Research Themes Presentation Title Here Subtitle
Presentation Title Here Subtitle