СИСТЕМА РЕГИСТРАЦИИ НОВЫХ ПРЕПАРАТОВ ДЛЯ IN VITRO ДИАГНОСТИКИ: СРАВНЕНИЕ РОССИЙСКОГО И МИРОВОГО ОПЫТА
- СИСТЕМА РЕГИСТРАЦИИ НОВЫХ ПРЕПАРАТОВ ДЛЯ IN VITRO ДИАГНОСТИКИ: СРАВНЕНИЕ РОССИЙСКОГО И МИРОВОГО ОПЫТА

Содержание
- 2. «Новый подход» к технической гармонизации Для медицинских изделий существуют три группы Директив, которые содержат исчерпывающие определения
- 3. Директивы устанавливают "существенные требования" к безопасности, проектированию и изготовлению ИМН, которым они должны отвечать при производстве
- 4. За оценку соответствия несут ответственность непосредственно изготовители или их уполномоченные представители, а также, более редко, специальные
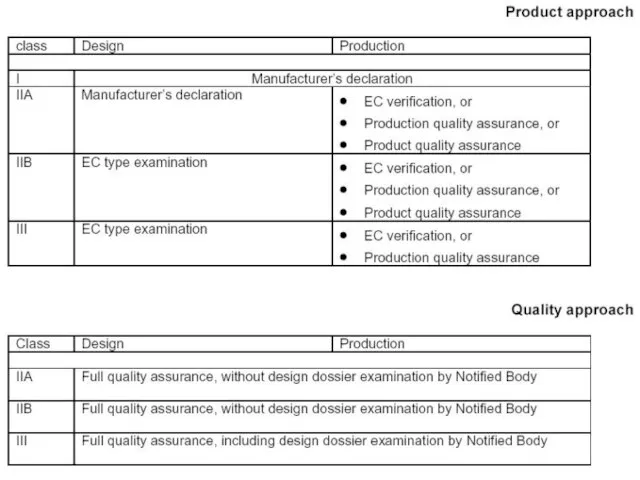
- 5. Директивы содержат процедуры оценки соответствия, которые зависят от типа изделий и типа имеющихся рисков. Кроме случаев
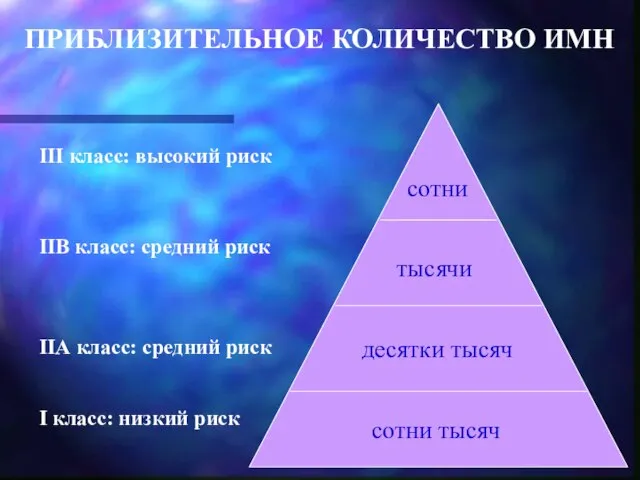
- 6. ПРИБЛИЗИТЕЛЬНОЕ КОЛИЧЕСТВО ИМН III класс: высокий риск IIВ класс: средний риск IIА класс: средний риск I
- 8. EU Directives The Medical Devices Directive (MDD) – применяется для всех медицинских изделий и аксессуаров кроме
- 9. EU Directives The In Vitro Diagnostics Directive (IVDD) – применяется для всех медицинских устройств (приборов) и
- 10. ‘Medical device` means any instrument, apparatus, appliance, material or other article, whether used alone or in
- 11. DIRECTIVE 98/79/EC on in vitro diagnostic medical devices Изделие медицинского назначения (ИМН) для диагностики in vitro
- 12. DIRECTIVE 98/79/EC on in vitro diagnostic medical devices Приложение II Список A - Изделия для определения
- 13. DIRECTIVE 98/79/EC on in vitro diagnostic medical devices Приложение II Список В - Изделия для определения
- 14. ANNEX X CE MARKING OF CONFORMITY The CE conformity marking shall consist of the initials ‘CE’
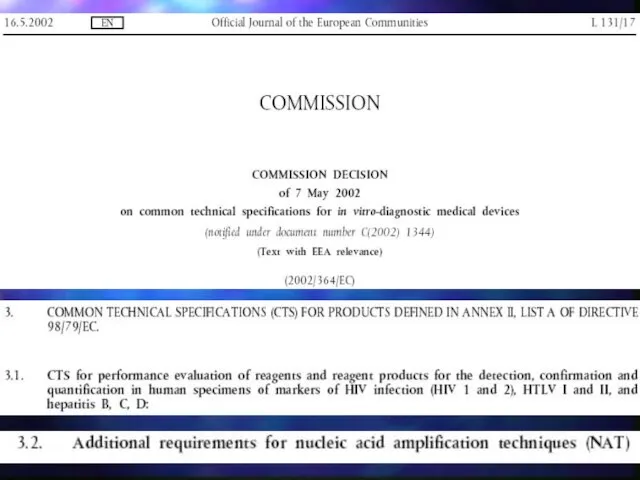
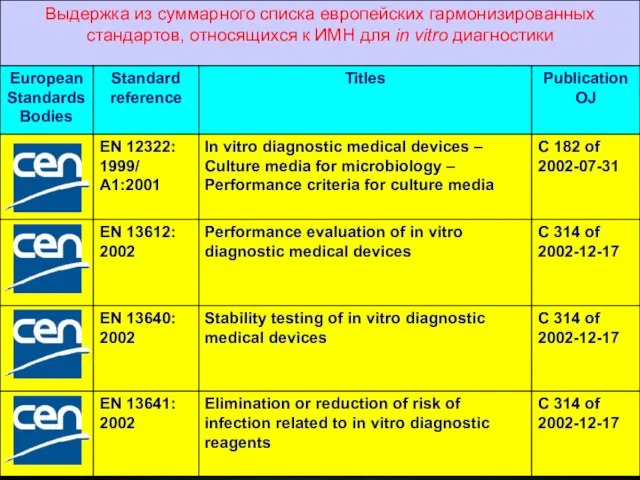
- 15. В отношение ИМН для in vitro диагностики приняты общие технические условия: Common Technical Specifications (CTS), устанавливающие
- 18. [Code of Federal Regulations] [Title 21, Volume 8] CHAPTER I- FOOD AND DRUG ADMINISTRATION, DEPARTMENT OF
- 19. Device Class and Regulatory Controls Class I. General Controls With Exemptions Without Exemptions Class II. General
- 20. Класс I. Общий контроль включает: Регистрацию предприятий (Establishment Registration, по Форме FDA 2891), которые обязаны регистрироваться
- 21. Класс II. Общий контроль и специальный контроль Специальный контроль может включать специальные требования к маркировке, обязательные
- 22. Класс III. Общий контроль и предпродажное одобрение/лицензирование (Premarket Approval) CBER регулирует медицинские изделия, участвующие в сборе,
- 23. The Global Harmonisation Task Force (GHTF) on medical devices (Целевая группа глобальной гармонизации обращения медицинских устройств)
- 24. Несоответствие правил обращения отечественной продукции требованиям международных стандартов - один из главных барьеров в попытках прорваться
- 25. Двухуровневая система технических регламентов и национальных стандартов. Первые обязательны для исполнения, для вторых установлен статус добровольных.
- 26. Национальные стандарты будут содержать дополнительные требования к качеству продукции (вместо ГОСТов, ОСТов, СНиПов, СанПиНов и т.д.)
- 27. Статья 12. Принципы стандартизации Стандартизация осуществляется в соответствии с принципами: добровольного применения стандартов; максимального учета при
- 28. Проект национального стандарта «Правила организации производства и контроля качества лекарственных средств» разработан Ассоциацией инженеров по контролю
- 29. На основе этих проектов в марте 2004 года техническим комитетом по стандартизации ТК 458 был одобрен
- 31. Закон «О медицинских изделиях» существует только в проекте. Сроки разработки технических регламентов и национальных стандартов для
- 32. Санитарно-эпидемиологические правила СП 3.3.2.1288-03 "Надлежащая практика производства медицинских иммунобиологических препаратов" Дата введения 25 июня 2003 г.
- 34. ЛЕКАРСТВЕННОЕ СРЕДСТВО (Medicinal product) Любое вещество или комбинация веществ, предназначенное для лечения или профилактики заболеваний у
- 35. Медицинские иммунобиологические препараты (МИБП) ранее регистрировались обособленно от лекарственных средств. Существовали разные ОСТы и РД. Сейчас
- 36. Медицинские иммунобиологические препараты (МИБП): препараты для лечения, профилактики и диагностики инфекционных и других заболеваний человека, приготовленные,
- 37. Санитарные правила СП 3.3.2.015-94 "Производство и контроль медицинских иммунобиологических препаратов для обеспечения их качества" (утв. постановлением
- 38. Средства in vitro диагностики инфекционных болезней в РФ рассматриваются на практике как лекарственные средства. Однако напрямую
- 39. Федеральный закон от 22 июня 1998 г. № 86-ФЗ «О лекарственных средствах» (с изменениями от 2
- 40. Федеральный закон от 22 июня 1998 г. № 86-ФЗ «О лекарственных средствах» (с изменениями от 2
- 41. Отраслевой стандарт ОСТ 42-510-98 "Правила организации производства и контроля качества лекарственных средств (GMP) (утв. Минздравом РФ
- 42. Отраслевой стандарт ОСТ 91500.05.001.00 "Стандарты качества лекарственных средств. Основные положения" (утвержден приказом Минздрава РФ от 1
- 43. Отраслевой стандарт ОСТ 91500.05.001.00 "Стандарты качества лекарственных средств. Основные положения" (утвержден приказом Минздрава РФ от 1
- 44. Диагностические средства неинвазивного применения по мере развития новых технологий все в большей степени становятся похожими на
- 45. ArrayTube System
- 47. Одновременные множественные количественные иммуноферментные анализы Хемилюминисценция 90 биочипов в кассете
- 49. Препараты для in vitro диагностики необходимо исключить из лекарственных средств и рассматривать в соответствии с международной
- 50. После редактирования ФСП с сопроводительными письмами следует по маршруту Фармкомитет > ГИСК > Предприятие > ГИСК
- 51. Требования к деталям оформления ФСП в последнее время регулярно и часто видоизменяются, в результате проект ФСП
- 52. Целесообразно отдать функцию утверждения НД в одни руки. ГИСКу им. Л.А. Тарасевича следует придать международно-признанный статус
- 53. Нормативная документация в соответствии с международными стандартами Введение автоматизированной компьютерной системы регистрации (оценки соответствия) НД Установление
- 55. Скачать презентацию
Слайд 2
«Новый подход»
к технической гармонизации
Для медицинских изделий существуют три группы Директив, которые содержат
«Новый подход»
к технической гармонизации
Для медицинских изделий существуют три группы Директив, которые содержат

Слайд 3Директивы устанавливают "существенные требования" к безопасности, проектированию и изготовлению ИМН, которым они
Директивы устанавливают "существенные требования" к безопасности, проектированию и изготовлению ИМН, которым они

Слайд 4За оценку соответствия несут ответственность непосредственно изготовители или их уполномоченные представители, а
За оценку соответствия несут ответственность непосредственно изготовители или их уполномоченные представители, а

Слайд 5Директивы содержат процедуры оценки соответствия, которые зависят от типа изделий и типа
Директивы содержат процедуры оценки соответствия, которые зависят от типа изделий и типа

Слайд 6ПРИБЛИЗИТЕЛЬНОЕ КОЛИЧЕСТВО ИМН
III класс: высокий риск
IIВ класс: средний риск
IIА класс: средний риск
I
ПРИБЛИЗИТЕЛЬНОЕ КОЛИЧЕСТВО ИМН
III класс: высокий риск
IIВ класс: средний риск
IIА класс: средний риск
I
Слайд 8
EU Directives
The Medical Devices Directive (MDD) – применяется для всех медицинских изделий
EU Directives
The Medical Devices Directive (MDD) – применяется для всех медицинских изделий

Слайд 9EU Directives
The In Vitro Diagnostics Directive (IVDD) – применяется для всех медицинских
EU Directives
The In Vitro Diagnostics Directive (IVDD) – применяется для всех медицинских

Слайд 10
‘Medical device` means any instrument, apparatus, appliance, material or other article, whether
‘Medical device` means any instrument, apparatus, appliance, material or other article, whether

Слайд 11
DIRECTIVE 98/79/EC on in vitro diagnostic medical devices
Изделие медицинского назначения (ИМН) для
DIRECTIVE 98/79/EC on in vitro diagnostic medical devices
Изделие медицинского назначения (ИМН) для

Слайд 12
DIRECTIVE 98/79/EC on in vitro diagnostic medical devices
Приложение II
Список A
- Изделия
DIRECTIVE 98/79/EC on in vitro diagnostic medical devices
Приложение II
Список A
- Изделия

Слайд 13
DIRECTIVE 98/79/EC on in vitro diagnostic medical devices
Приложение II
Список В
- Изделия
DIRECTIVE 98/79/EC on in vitro diagnostic medical devices
Приложение II
Список В
- Изделия

Слайд 14ANNEX X
CE MARKING OF CONFORMITY
The CE conformity marking shall consist of the
ANNEX X
CE MARKING OF CONFORMITY
The CE conformity marking shall consist of the

Слайд 15В отношение ИМН для in vitro диагностики приняты общие технические условия: Common
В отношение ИМН для in vitro диагностики приняты общие технические условия: Common

Слайд 18[Code of Federal Regulations]
[Title 21, Volume 8]
CHAPTER I- FOOD AND DRUG ADMINISTRATION,
[Code of Federal Regulations]
[Title 21, Volume 8]
CHAPTER I- FOOD AND DRUG ADMINISTRATION,
![[Code of Federal Regulations] [Title 21, Volume 8] CHAPTER I- FOOD AND](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/391964/slide-17.jpg)
Слайд 19Device Class and Regulatory Controls
Class I. General Controls
With Exemptions
Without
Device Class and Regulatory Controls
Class I. General Controls
With Exemptions
Without

Слайд 20Класс I. Общий контроль включает:
Регистрацию предприятий (Establishment Registration, по Форме FDA 2891),
Класс I. Общий контроль включает:
Регистрацию предприятий (Establishment Registration, по Форме FDA 2891),

Слайд 21Класс II. Общий контроль и специальный контроль
Специальный контроль может включать специальные требования
Класс II. Общий контроль и специальный контроль
Специальный контроль может включать специальные требования

Слайд 22Класс III. Общий контроль и предпродажное одобрение/лицензирование (Premarket Approval)
CBER регулирует медицинские изделия,
Класс III. Общий контроль и предпродажное одобрение/лицензирование (Premarket Approval)
CBER регулирует медицинские изделия,

Слайд 23The Global Harmonisation Task Force (GHTF) on medical devices
(Целевая группа глобальной
The Global Harmonisation Task Force (GHTF) on medical devices (Целевая группа глобальной

Слайд 24Несоответствие правил обращения отечественной продукции требованиям международных стандартов - один из главных
Несоответствие правил обращения отечественной продукции требованиям международных стандартов - один из главных

Слайд 25Двухуровневая система технических регламентов и национальных стандартов. Первые обязательны для исполнения, для
Двухуровневая система технических регламентов и национальных стандартов. Первые обязательны для исполнения, для

Слайд 26Национальные стандарты будут содержать дополнительные требования к качеству продукции (вместо ГОСТов, ОСТов,
Национальные стандарты будут содержать дополнительные требования к качеству продукции (вместо ГОСТов, ОСТов,

Слайд 27Статья 12. Принципы стандартизации
Стандартизация осуществляется в соответствии с принципами:
добровольного применения стандартов;
максимального учета
Статья 12. Принципы стандартизации
Стандартизация осуществляется в соответствии с принципами:
добровольного применения стандартов;
максимального учета

Слайд 28Проект национального стандарта «Правила организации производства и контроля качества лекарственных средств» разработан
Проект национального стандарта «Правила организации производства и контроля качества лекарственных средств» разработан

Слайд 29На основе этих проектов в марте 2004 года техническим комитетом по стандартизации
На основе этих проектов в марте 2004 года техническим комитетом по стандартизации


Слайд 31Закон «О медицинских изделиях» существует только в проекте.
Сроки разработки технических регламентов и
Закон «О медицинских изделиях» существует только в проекте.
Сроки разработки технических регламентов и

Слайд 32Санитарно-эпидемиологические правила
СП 3.3.2.1288-03
"Надлежащая практика производства медицинских иммунобиологических препаратов"
Дата введения 25 июня
Санитарно-эпидемиологические правила
СП 3.3.2.1288-03
"Надлежащая практика производства медицинских иммунобиологических препаратов"
Дата введения 25 июня


Слайд 34ЛЕКАРСТВЕННОЕ СРЕДСТВО (Medicinal product)
Любое вещество или комбинация веществ, предназначенное для лечения или
ЛЕКАРСТВЕННОЕ СРЕДСТВО (Medicinal product)
Любое вещество или комбинация веществ, предназначенное для лечения или

Слайд 35Медицинские иммунобиологические препараты (МИБП) ранее регистрировались обособленно от лекарственных средств.
Существовали разные ОСТы
Медицинские иммунобиологические препараты (МИБП) ранее регистрировались обособленно от лекарственных средств.
Существовали разные ОСТы

Слайд 36Медицинские иммунобиологические препараты (МИБП):
препараты для лечения, профилактики и диагностики инфекционных и
Медицинские иммунобиологические препараты (МИБП):
препараты для лечения, профилактики и диагностики инфекционных и

Слайд 37Санитарные правила СП 3.3.2.015-94
"Производство и контроль медицинских иммунобиологических
препаратов для обеспечения их качества"
(утв.
"Производство и контроль медицинских иммунобиологических
препаратов для обеспечения их качества"
(утв.

Слайд 38Средства in vitro диагностики инфекционных болезней в РФ рассматриваются на практике как
Средства in vitro диагностики инфекционных болезней в РФ рассматриваются на практике как

Слайд 39Федеральный закон от 22 июня 1998 г. № 86-ФЗ
«О лекарственных средствах»
(с изменениями
Федеральный закон от 22 июня 1998 г. № 86-ФЗ «О лекарственных средствах» (с изменениями

Слайд 40Федеральный закон от 22 июня 1998 г. № 86-ФЗ
«О лекарственных средствах»
(с изменениями
Федеральный закон от 22 июня 1998 г. № 86-ФЗ «О лекарственных средствах» (с изменениями

Слайд 41Отраслевой стандарт ОСТ 42-510-98
"Правила организации производства и контроля качества лекарственных средств (GMP)
(утв.
Отраслевой стандарт ОСТ 42-510-98
"Правила организации производства и контроля качества лекарственных средств (GMP)
(утв.

Слайд 42Отраслевой стандарт ОСТ 91500.05.001.00
"Стандарты качества лекарственных средств. Основные положения"
(утвержден приказом Минздрава РФ
Отраслевой стандарт ОСТ 91500.05.001.00
"Стандарты качества лекарственных средств. Основные положения"
(утвержден приказом Минздрава РФ

Слайд 43Отраслевой стандарт ОСТ 91500.05.001.00
"Стандарты качества лекарственных средств. Основные положения"
(утвержден приказом Минздрава РФ
Отраслевой стандарт ОСТ 91500.05.001.00
"Стандарты качества лекарственных средств. Основные положения"
(утвержден приказом Минздрава РФ

Слайд 44Диагностические средства неинвазивного применения по мере развития новых технологий все в большей
Диагностические средства неинвазивного применения по мере развития новых технологий все в большей

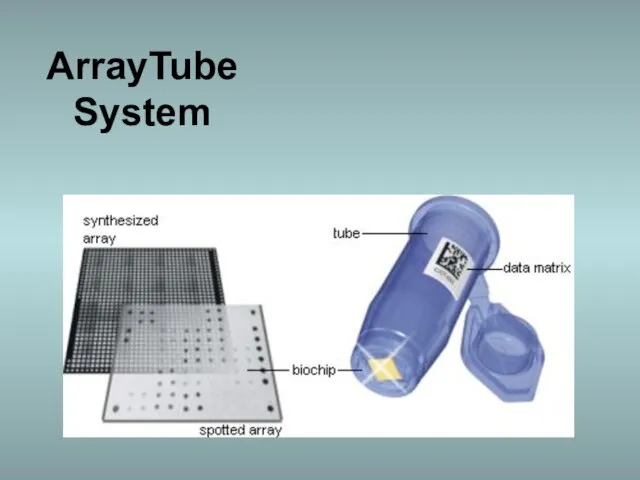
Слайд 45ArrayTube System
ArrayTube System

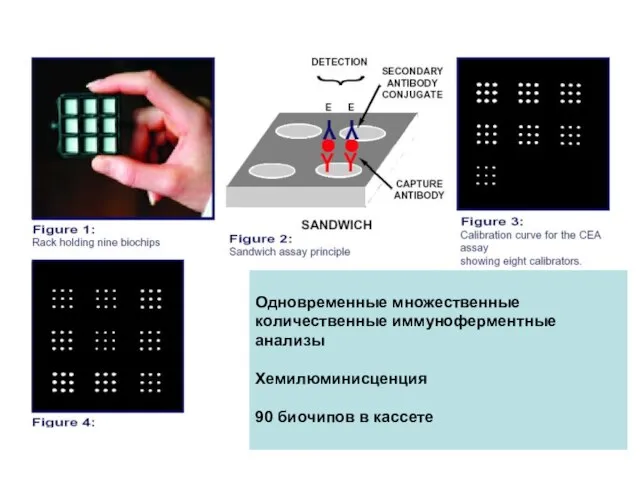
Слайд 47Одновременные множественные количественные иммуноферментные анализы
Хемилюминисценция
90 биочипов в кассете
Хемилюминисценция
90 биочипов в кассете

Слайд 49Препараты для in vitro диагностики необходимо исключить из лекарственных средств и рассматривать
Препараты для in vitro диагностики необходимо исключить из лекарственных средств и рассматривать

Слайд 50После редактирования ФСП с сопроводительными письмами следует по маршруту Фармкомитет > ГИСК
После редактирования ФСП с сопроводительными письмами следует по маршруту Фармкомитет > ГИСК

Слайд 51Требования к деталям оформления ФСП в последнее время регулярно и часто видоизменяются,
Требования к деталям оформления ФСП в последнее время регулярно и часто видоизменяются,

Слайд 52Целесообразно отдать функцию утверждения НД в одни руки.
ГИСКу им. Л.А. Тарасевича следует придать международно-признанный
Целесообразно отдать функцию утверждения НД в одни руки.
ГИСКу им. Л.А. Тарасевича следует придать международно-признанный

Слайд 53Нормативная документация в соответствии с международными стандартами
Введение автоматизированной компьютерной системы регистрации (оценки
Нормативная документация в соответствии с международными стандартами
Введение автоматизированной компьютерной системы регистрации (оценки

 Анонимный опрос учащихсяМОУ Усовская сош5-8 классов«Личный опыт школьника»
Анонимный опрос учащихсяМОУ Усовская сош5-8 классов«Личный опыт школьника» Гимнастика. Термин
Гимнастика. Термин Шаблон для инициативного проекта
Шаблон для инициативного проекта Перпендикулярность прямой и плоскости
Перпендикулярность прямой и плоскости Презентация на тему Организация и техника внешнеэкономических операций по купле-продаже лицензий и по международному обмену инж
Презентация на тему Организация и техника внешнеэкономических операций по купле-продаже лицензий и по международному обмену инж ДЕРЕВЯННАЯ МОЗАИКА
ДЕРЕВЯННАЯ МОЗАИКА Доклад руководителя Федерального дорожного агентства Чабунина Анатолия Михайловича
Доклад руководителя Федерального дорожного агентства Чабунина Анатолия Михайловича Презентация тема 2 (2)
Презентация тема 2 (2) бази даних
бази даних цветущий сад оригами
цветущий сад оригами Кофе латте
Кофе латте Избирательный процесс
Избирательный процесс Статистическое наблюдение по вопросам использования населением информационных технологий и сетей
Статистическое наблюдение по вопросам использования населением информационных технологий и сетей «Поисковое продвижение сайтов»
«Поисковое продвижение сайтов» Презентация экспериментальной площадки по программе дополнительного образования детей старшего дошкольного возраста «Внесем в
Презентация экспериментальной площадки по программе дополнительного образования детей старшего дошкольного возраста «Внесем в  Презентация на тему Испарение. Насыщенный и ненасыщенный пар
Презентация на тему Испарение. Насыщенный и ненасыщенный пар  Unit 11-5. Презентация
Unit 11-5. Презентация Народные промыслы России
Народные промыслы России Марийская вышивка
Марийская вышивка Открытое факультативное занятие «Здоровый образ жизни» во 2 «Б» классе ГУО «Средняя школа №34 г. Могилева» Учитель начальных к
Открытое факультативное занятие «Здоровый образ жизни» во 2 «Б» классе ГУО «Средняя школа №34 г. Могилева» Учитель начальных к Проблемы выявления и диагностики ранних стадий хронической болезни почек
Проблемы выявления и диагностики ранних стадий хронической болезни почек Свойства производной. Построение графиков функций
Свойства производной. Построение графиков функций Omapalvelu. Lisäselvityspyyntö
Omapalvelu. Lisäselvityspyyntö Презентация на тему Музыкальная живопись и живописная музыка (5 класс)
Презентация на тему Музыкальная живопись и живописная музыка (5 класс) Пекарь кондитер
Пекарь кондитер Использование электромагнитов
Использование электромагнитов Орнамент. 1 класс
Орнамент. 1 класс Мое представление о семье
Мое представление о семье