- Введение в организацию белковой структуры и молекулярное моделирование

Содержание
- 2. Проблематика самоорганизации структуры белка “Предсказание трехмерной структуры белка по его аминокислотной последовательности.” “Каким образом белковая молекула
- 3. Белки могут сворачиваться в свои третичные структуры спонтанно В растворах белковые молекулы самостоятельно организуются в трехмерные
- 4. Парадокс Левинтала Можем предположить, что для каждой аминокислоты существует три варианта ее состояния в белке (α-спираль,
- 5. Почему “Самоорганизация структуры белка” так важна? Белки играют важные роли в живых организмах. Некоторые белки напрямую
- 6. Почему проблема “Самоорганизации структуры белка” так сложна? С точки зрения компьютерного моделирования, Сложно моделировать весь процесс
- 7. Молекулярная Динамика (MD) В модели молекулярной динамики, мы имитируем движение атомов как функцию от времени на
- 8. Интегрирование с использованием метода конечной разности Положения в моменты времени (t + Δt ) и (t
- 9. Силы, участвующие в процессе самоорганизации белковой молекулы Электростатические взаимодействия Силы Ван дер Ваальса Водородные связи Гидрофобные
- 10. Функции от составляющих сил, рассчитываемые в Молекулярном Моделировании Электростатическая сила Водородная связь Сила Ван дер Ваальса
- 11. Система для Моделирования с помощью Молекулярной Динамики Без молекул воды С молекулами воды Кол-во атомов: 304
- 12. MD требует колоссальных вычислительных мощностей Квант времени в методе MD (Δt) ограничен 1 фсек (10-15 сек).
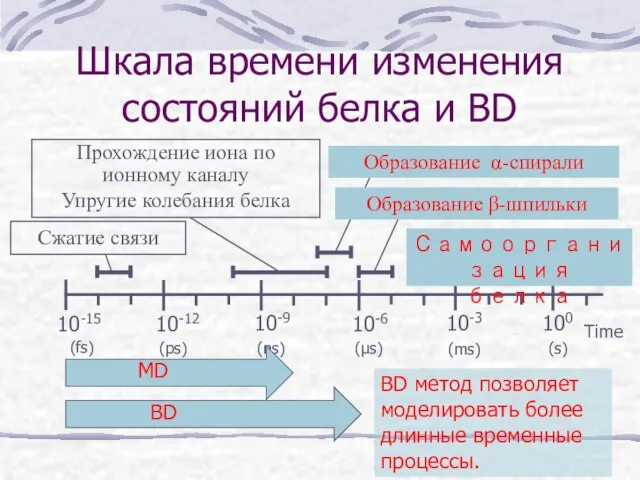
- 13. Шкала времени изменения состояний белка и MD Время Сжатие связи Прохождение иона по ионному каналу Упругие
- 14. Гораздо быстрее, Гораздо больше! Специальные компьютеры Расчет свободных взаимодействий (не химических связей) обеспечивается с использованием специальных
- 15. Броуновская Динамика (BD) Динамический вклад растворителя представлен в виде рассеянного случайного воздействия (Открытие Эйнштейна в 1905).
- 16. Система для BD Моделирования Без молекул воды С молекулами воды Кол-во атомов: 304 Кол-во атомов: 304
- 17. Алгоритм BD Уравнение Ланжевина может быть выражено так: Здесь, ri и mi отражают соответственно положение и
- 18. Вычислительное время BD †MTS(Multiple time step) алгоритм: Этот метод позволяет уменьшить необходимость расчетов самой тяжелой части
- 19. Моделирование образования α-спирали с помощью BD Доля возможных в природе связей Время расчета (нсек) 0 300
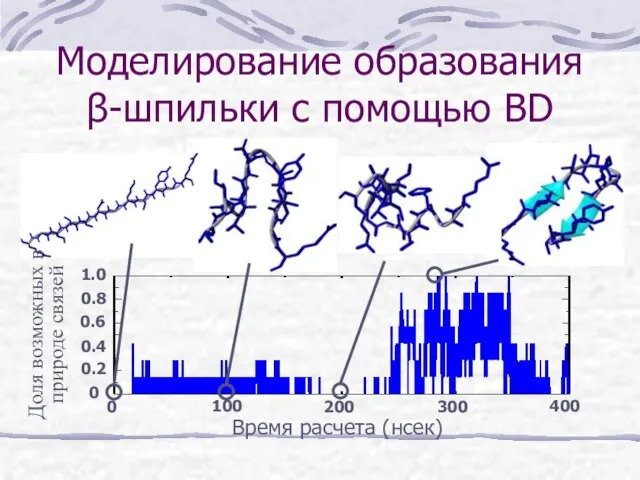
- 20. Моделирование образования β-шпильки с помощью BD Доля возможных в природе связей Время расчета (нсек) 0 300
- 21. Шкала времени изменения состояний белка и BD Time BD метод позволяет моделировать более длинные временные процессы.
- 23. Скачать презентацию
Слайд 2Проблематика самоорганизации структуры белка
“Предсказание трехмерной структуры белка по его аминокислотной последовательности.”
“Каким образом
Проблематика самоорганизации структуры белка
“Предсказание трехмерной структуры белка по его аминокислотной последовательности.”
“Каким образом

Слайд 3Белки могут сворачиваться в свои третичные структуры спонтанно
В растворах белковые молекулы самостоятельно
Белки могут сворачиваться в свои третичные структуры спонтанно
В растворах белковые молекулы самостоятельно

Слайд 4Парадокс Левинтала
Можем предположить, что для каждой аминокислоты существует три варианта ее состояния
Парадокс Левинтала
Можем предположить, что для каждой аминокислоты существует три варианта ее состояния

Слайд 5Почему “Самоорганизация структуры белка” так важна?
Белки играют важные роли в живых организмах.
Некоторые
Почему “Самоорганизация структуры белка” так важна?
Белки играют важные роли в живых организмах.
Некоторые

Слайд 6Почему проблема “Самоорганизации структуры белка” так сложна?
С точки зрения компьютерного моделирования,
Сложно моделировать
Почему проблема “Самоорганизации структуры белка” так сложна?
С точки зрения компьютерного моделирования,
Сложно моделировать

Слайд 7Молекулярная Динамика (MD)
В модели молекулярной динамики, мы имитируем движение атомов как функцию
Молекулярная Динамика (MD)
В модели молекулярной динамики, мы имитируем движение атомов как функцию

Слайд 8Интегрирование с использованием метода конечной разности
Положения в моменты времени (t + Δt
Интегрирование с использованием метода конечной разности
Положения в моменты времени (t + Δt

Слайд 9Силы, участвующие в процессе самоорганизации белковой молекулы
Электростатические взаимодействия
Силы Ван дер Ваальса
Водородные связи
Гидрофобные
Силы, участвующие в процессе самоорганизации белковой молекулы
Электростатические взаимодействия
Силы Ван дер Ваальса
Водородные связи
Гидрофобные

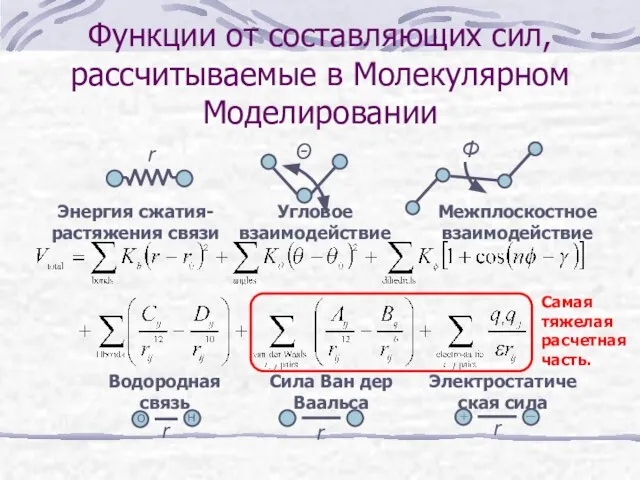
Слайд 10Функции от составляющих сил, рассчитываемые в Молекулярном Моделировании
Электростатическая сила
Водородная связь
Сила Ван дер
Функции от составляющих сил, рассчитываемые в Молекулярном Моделировании
Электростатическая сила
Водородная связь
Сила Ван дер
Слайд 11Система для Моделирования с помощью Молекулярной Динамики
Без молекул воды
С молекулами воды
Кол-во атомов:
Система для Моделирования с помощью Молекулярной Динамики
Без молекул воды
С молекулами воды
Кол-во атомов:

Слайд 12MD требует колоссальных вычислительных мощностей
Квант времени в методе MD (Δt) ограничен 1
MD требует колоссальных вычислительных мощностей
Квант времени в методе MD (Δt) ограничен 1

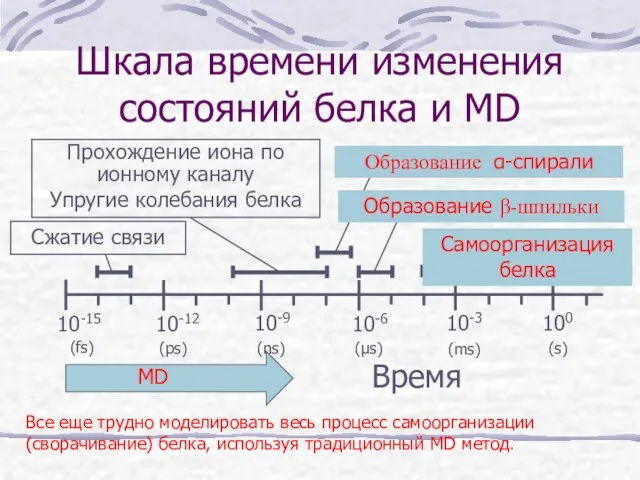
Слайд 13Шкала времени изменения состояний белка и MD
Время
Сжатие связи
Прохождение иона по ионному каналу
Упругие
Шкала времени изменения состояний белка и MD
Время
Сжатие связи
Прохождение иона по ионному каналу
Упругие
Слайд 14Гораздо быстрее, Гораздо больше!
Специальные компьютеры
Расчет свободных взаимодействий (не химических связей) обеспечивается с
Гораздо быстрее, Гораздо больше!
Специальные компьютеры
Расчет свободных взаимодействий (не химических связей) обеспечивается с

Слайд 15Броуновская Динамика (BD)
Динамический вклад растворителя представлен в виде рассеянного случайного воздействия (Открытие
Броуновская Динамика (BD)
Динамический вклад растворителя представлен в виде рассеянного случайного воздействия (Открытие

Слайд 16Система для BD Моделирования
Без молекул воды
С молекулами воды
Кол-во атомов: 304
Кол-во атомов: 304 +
Система для BD Моделирования
Без молекул воды
С молекулами воды
Кол-во атомов: 304
Кол-во атомов: 304 +

Слайд 17Алгоритм BD
Уравнение Ланжевина может быть выражено так:
Здесь, ri и mi отражают соответственно
Алгоритм BD
Уравнение Ланжевина может быть выражено так: Здесь, ri и mi отражают соответственно

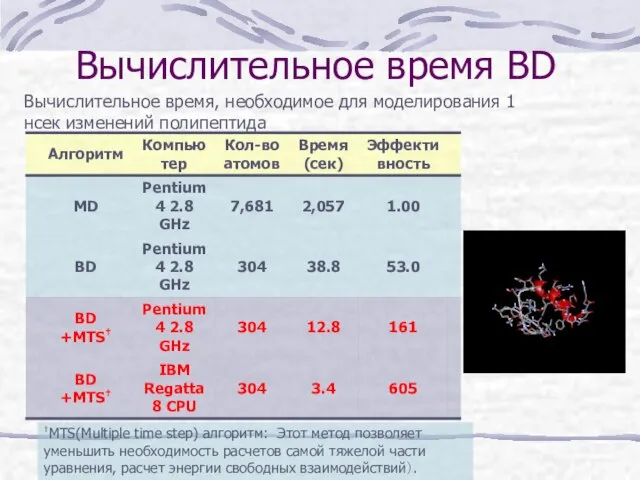
Слайд 18Вычислительное время BD
†MTS(Multiple time step) алгоритм: Этот метод позволяет уменьшить необходимость расчетов самой
Вычислительное время BD
†MTS(Multiple time step) алгоритм: Этот метод позволяет уменьшить необходимость расчетов самой
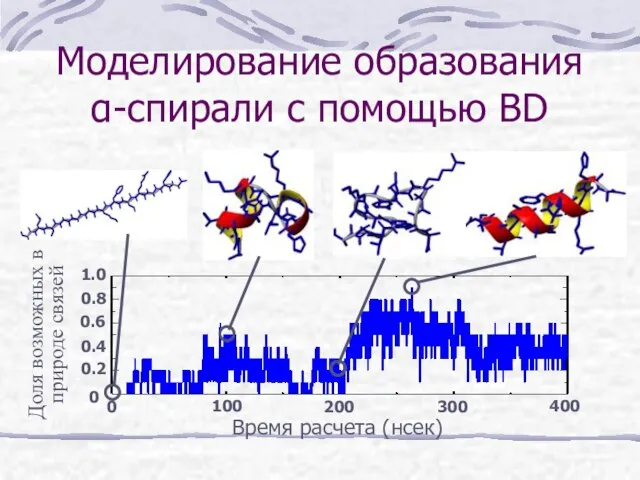
Слайд 19Моделирование образования α-спирали с помощью BD
Доля возможных в природе связей
Время расчета (нсек)
0
300
200
100
400
0
0.2
0.4
0.6
0.8
1.0
Моделирование образования α-спирали с помощью BD
Доля возможных в природе связей
Время расчета (нсек)
0
300
200
100
400
0
0.2
0.4
0.6
0.8
1.0
Слайд 20Моделирование образования
β-шпильки с помощью BD
Доля возможных в природе связей
Время расчета (нсек)
0
300
200
100
400
0
0.2
0.4
0.6
0.8
1.0
Моделирование образования
β-шпильки с помощью BD
Доля возможных в природе связей
Время расчета (нсек)
0
300
200
100
400
0
0.2
0.4
0.6
0.8
1.0
Слайд 21Шкала времени изменения состояний белка и BD
Time
BD метод позволяет моделировать более длинные
Шкала времени изменения состояний белка и BD
Time
BD метод позволяет моделировать более длинные
 КОТЕЛЬНАЯ 4 Х 15 МВт
КОТЕЛЬНАЯ 4 Х 15 МВт Управленческие прогнозы в производстве и маркетинге. Случай подозрительного потребителя
Управленческие прогнозы в производстве и маркетинге. Случай подозрительного потребителя Нательные кресты XI – конца XIX вв. в экспозиции музея им. 1000-летия Брянска
Нательные кресты XI – конца XIX вв. в экспозиции музея им. 1000-летия Брянска Иерархия потребностей по Маслоу
Иерархия потребностей по Маслоу What time is it
What time is it Стили и направления танца
Стили и направления танца Портрет в музыке и живописи
Портрет в музыке и живописи Абажур. Уроки технологии
Абажур. Уроки технологии Прочитайте отрывок из труда Н.И. Костомарова об отношении к реформам населения в XVII в.? «…а мы от разумных людей слыхали: которая зе
Прочитайте отрывок из труда Н.И. Костомарова об отношении к реформам населения в XVII в.? «…а мы от разумных людей слыхали: которая зе Введение в китайский язык
Введение в китайский язык Отчёт о работе ШМО учителей русского языка и литературы МОУ «ООСШ№2» за 2010 – 2011 уч. год
Отчёт о работе ШМО учителей русского языка и литературы МОУ «ООСШ№2» за 2010 – 2011 уч. год Тире и двоеточие в бессоюзном сложном предложении
Тире и двоеточие в бессоюзном сложном предложении Кошки - особый и таинственный мир
Кошки - особый и таинственный мир Стили архитектуры (Проверочная работа № 2)
Стили архитектуры (Проверочная работа № 2) ТАС Инвест. Процедура заключения договора страхования
ТАС Инвест. Процедура заключения договора страхования Живое электричество Проектная группа 10-х классов МОУ «Лицей №10» Пермь, 2009
Живое электричество Проектная группа 10-х классов МОУ «Лицей №10» Пермь, 2009 Расшифровка бодиграфа. Консультации по дизайну человека
Расшифровка бодиграфа. Консультации по дизайну человека Проблемы и принципы открытого доступа к научной информации
Проблемы и принципы открытого доступа к научной информации СРАВНИТЕЛЬНАЯ ЭФФЕКТИВНОСТЬ КВАМАТЕЛА И ОМЕПРАЗОЛА ПРИ ЛЕЧЕНИИ БОЛЬНЫХ ХРОНИЧЕСКИМ ПАНКРЕАТИТОМ
СРАВНИТЕЛЬНАЯ ЭФФЕКТИВНОСТЬ КВАМАТЕЛА И ОМЕПРАЗОЛА ПРИ ЛЕЧЕНИИ БОЛЬНЫХ ХРОНИЧЕСКИМ ПАНКРЕАТИТОМ Диодная система Clearlight. Золотой стандарт лазерной эпиляции
Диодная система Clearlight. Золотой стандарт лазерной эпиляции Воздушно-дуговая резка
Воздушно-дуговая резка ПОВЫШЕНИЕ КАЧЕСТВА ОБРАЗОВАНИЯ ЧЕРЕЗ РЕАЛИЗАЦИЮ КОМПЕТЕНТНОСТНОГО И СИСТЕМНО– ДЕЯТЕЛЬНОСТНОГО ПОДХОДА В РАБОТЕ ПЕДАГОГА
ПОВЫШЕНИЕ КАЧЕСТВА ОБРАЗОВАНИЯ ЧЕРЕЗ РЕАЛИЗАЦИЮ КОМПЕТЕНТНОСТНОГО И СИСТЕМНО– ДЕЯТЕЛЬНОСТНОГО ПОДХОДА В РАБОТЕ ПЕДАГОГА Классификация химических реакций 11 класс
Классификация химических реакций 11 класс Поверхности. Определение и задание на чертеже
Поверхности. Определение и задание на чертеже Миграция и денежные переводы эмигрантовСтраны Восточной Европы и бывшего СССРВсемирный банкРегион Европы и Центральной Азии
Миграция и денежные переводы эмигрантовСтраны Восточной Европы и бывшего СССРВсемирный банкРегион Европы и Центральной Азии г.Санкт-Петербург Школа №557
г.Санкт-Петербург Школа №557 Eurocopter EC 130 B4
Eurocopter EC 130 B4 1С-Рейтинг: Больничная аптекадля 1С:Предприятие 8.2Учет лекарственных средств и аптечных товаров в организациях здравоохранения
1С-Рейтинг: Больничная аптекадля 1С:Предприятие 8.2Учет лекарственных средств и аптечных товаров в организациях здравоохранения