- Хромосомные мутации (аберрации)

Содержание
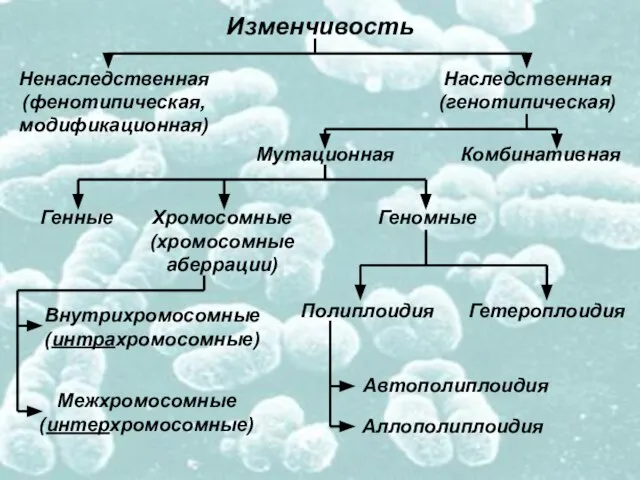
- 2. Изменчивость Ненаследственная (фенотипическая, модификационная) Наследственная (генотипическая) Мутационная Комбинативная Генные Хромосомные (хромосомные аберрации) Геномные Полиплоидия Гетероплоидия Автополиплоидия
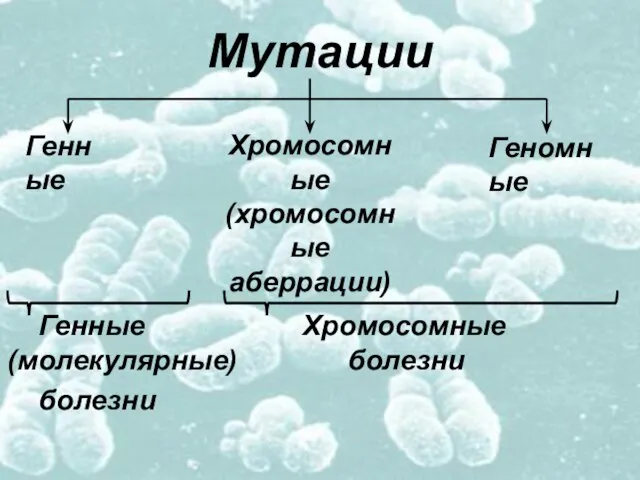
- 3. Мутации Генные Хромосомные (молекулярные) болезни болезни Генные Хромосомные (хромосомные аберрации) Геномные
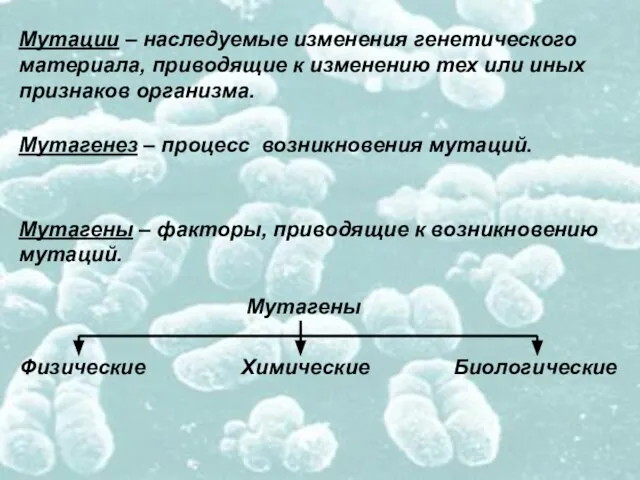
- 4. Мутации – наследуемые изменения генетического материала, приводящие к изменению тех или иных признаков организма. Мутагенез –
- 5. Хромосомные болезни – большая группа врожденных наследственных болезней, клинически характеризующихся множественными врожденными пороками развития. В их
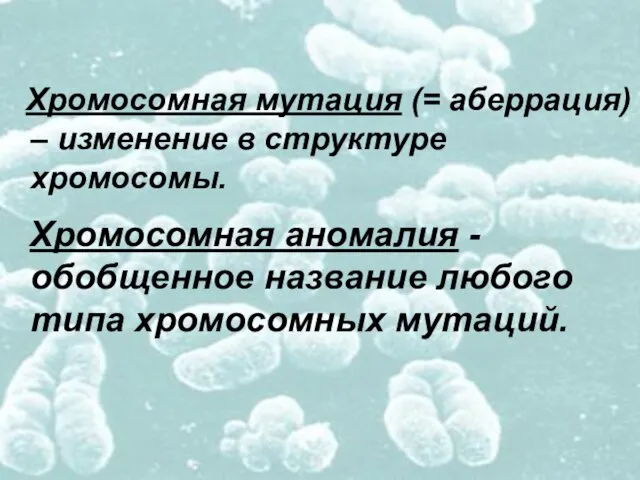
- 6. Хромосомная аномалия - обобщенное название любого типа хромосомных мутаций. Хромосомная мутация (= аберрация) – изменение в
- 7. Основная причина возникновения различных хромосомных мутаций – разрывы хромосом и хроматид и воссоединения в новых сочетаниях.
- 8. Хромосомные аберрации Внутрихромосомные (=интрахромосомные) Межхромосомные (=интерхромосомные) Делеция Терминальная Срединная Потеря теломерных участков Дупликация Инверсия Транспозиция Транслокации
- 9. 1. Делеция а) Терминальная Deficiency (=концевая) (потеря концевых участков) Возможна при одиночном разрыве.
- 10. 1. Делеция б) Срединная ( = интерстициальная ) Возможна при двойном разрыве.
- 11. 1. Делеция в) Образование кольцевой хромосомы Возможна при потере теломерных участков (в случае концевых делеций в
- 12. 2. Дупликация. Удвоение участка хромосомы.
- 13. 3. Инверсия Возможна при двойном разрыве и последующем повороте образовавшегося участка на 180 градусов. Инверсия -
- 14. 4. Транспозиция Возможна при тройных разрывах. Представляет собой перенос участка хромосомы на другое место на той
- 15. Транслокации а) Реципрокные ( = взаимные = сбалансированные). Две негомологичные хромосомы обмениваются участками .
- 16. Транслокации б) Нереципрокные ( = невзаимные = собственно транслокации). Перенос участка с одной негомологичной хромосомы (или
- 17. Транслокации в) Робертсоновские Две негомологичные хромосомы объединяются в одну . (Разрыв происходит в области центромер). Чаще
- 18. Реципрокная транслокация (1-3) + Терминальная делеция (9) : Кариотип.
- 19. Робертсоновская транслокация (13;14). Диагноз: невынашивание беременности. Кариотип: 45,XX, rob t (13;14)
- 20. Заболевания, вызываемые хромосомными аберрациями
- 21. Синдром Лежьена ( = Синдром «Кошачьего крика» ). Кариотип: 46,ХХ(ХY),B5p- Частота (у новорожденных): 1:45000 – 1:50000
- 22. Синдром Лежьена ( = Синдром «Кошачьего крика» ). Возможны и другие цитогенетические варианты: Кольцевая хромосома 5
- 23. Синдром Лежьена ( = Синдром «Кошачьего крика» ). Клиника: Монотонный или резкий, слабый или высокий крик,
- 24. Синдром Лежьена ( = Синдром «Кошачьего крика»). Выраженные признаки синдрома Маловыраженные признаки
- 25. Синдром Лежьена ( = Синдром «Кошачьего крика»).
- 26. Синдром Лежьена ( = Синдром «Кошачьего крика»).
- 27. Синдром Вольфа-Хиршхорна Кариотип: 46,ХХ(ХY), В4р- Обусловлен делецией короткого плеча хромосомы 4. За симптомокомплекс ответственен сегмент 4р16
- 28. Синдром Вольфа-Хиршхорна Клиника (по Н.П.Бочкову) : Микроцефалия; Клювовидный нос; Гипертелоризм; Эпикант; Аномальные ушные раковины; Расщелины верхней
- 29. Синдром Вольфа-Хиршхорна
- 30. Синдром де Груши (Синдром, обусловленный делецией короткого плеча 18 хромосомы) Частота: 1:60 000 Обусловлен делецией, происшедшей
- 31. Синдром де Груши (Синдром, обусловленный делецией короткого плеча 18 хромосомы) Клиника: Низкорослость; Маленькая окружность черепа без
- 32. Синдром де Груши (Синдром, обусловленный делецией короткого плеча 18 хромосомы)
- 33. Синдром 18q- Частота: 1:60 000 Обследовано 25 больных. Продолжительность жизни составляет 2,5 – 5 лет. Но
- 34. Синдром 18q- Клиника: Микроцефалия; Рот – маленький ( рот «карпа» ); Косоглазие, глаукома, атрофия зрительного нерва;
- 35. Дисгенезия гонад 46, ХХ р- 46, ХХ q- Потеря плеча Х-хромосомы вызывает признаки, сходные с теми,
- 36. Дисгенезия гонад Недоразвитие вторичных половых признаков.
- 37. Синдром «Кошачьего глаза» Синдром частичной трисомии хромосомы 22 Относится к особой группе заболеваний – наследственным синдромам
- 38. Синдром «Кошачьего глаза» Синдром частичной трисомии хромосомы 22 Описан в 1978 году. Встречается редко. Частота его
- 39. Синдром «Кошачьего глаза» Синдром частичной трисомии хромосомы 22 Клиника: Вертикальная колобома радужки («Кошачий глаз»); Атрезия ануса;
- 40. Синдром «Кошачьего глаза» Синдром частичной трисомии хромосомы 22
- 41. Синдром частичной трисомии по короткому плечу хромосомы 9 Обследовано более 200 больных. Является результатом несбалансированных транслокаций,
- 42. Синдром частичной трисомии по короткому плечу хромосомы 9 Клиника: Задержка роста; Умственная отсталость; Антимонголоидный разрез глаз;
- 43. Синдром частичной трисомии по короткому плечу хромосомы 9 Жизненный прогноз сравнительно благоприятный. Больные доживают до пожилого
- 44. Синдром частичной трисомии по короткому плечу хромосомы 9 Характерные признаки
- 45. Синдром Орбели (частичной моносомии 13q-) Частота 1:100 000 Обусловлен образованием на длинном плече хромосомы 13 терминальной
- 46. Синдром Орбели (частичной моносомии 13q-) Клиника: Лицо асимметричное Широкая, выступающая спинка носа – «Греческий профиль» Эпикант
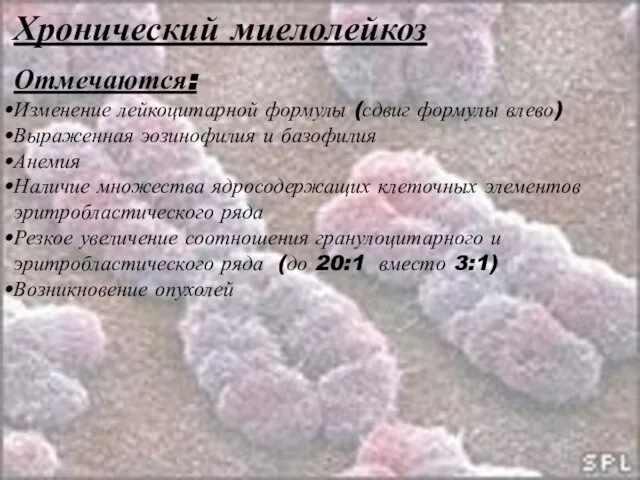
- 47. Хронический миелолейкоз У 95% больных в опухолевых клетках обнаруживается укороченная 22-я «Филадельфийская» хромосома, образующаяся в результате
- 48. Хронический миелолейкоз Отмечаются: Изменение лейкоцитарной формулы (сдвиг формулы влево) Выраженная эозинофилия и базофилия Анемия Наличие множества
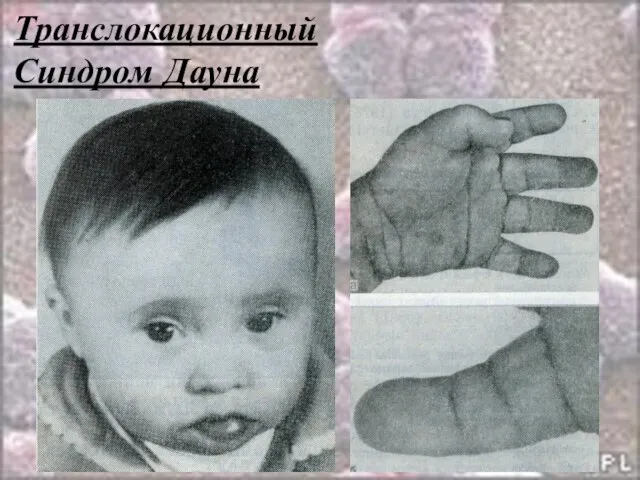
- 49. Транслокационный Синдром Дауна 46,ХХ(ХY),G21+/2,9,13-15,21-22t Примерно 3-4% больных с синдромом Дауна имеют транслокационную форму трисомии по типу
- 50. Транслокационный Синдром Дауна Клиника: Микроцефалия Умственная отсталость Голова уменьшенных размеров, череп круглый, затылок плоский Лицо плоское,
- 51. Транслокационный Синдром Дауна
- 52. Транслокационный Синдром Дауна Дети разного возраста с характерными чертами синдрома Дауна
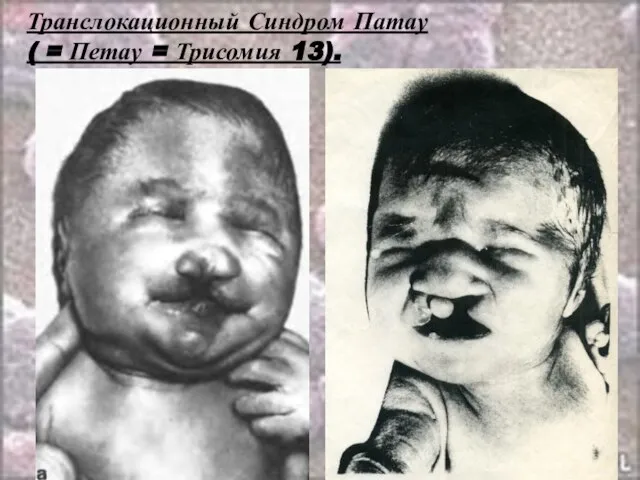
- 53. Транслокационный Синдром Патау ( = Петау = Трисомия 13). Кариотип: 46,ХХ(ХY),D 13+/2,9,13-15,21-22 t Соотношение полов близко
- 54. Транслокационный Синдром Патау ( = Петау = Трисомия 13). В связи с тяжелыми врожденными пороками развития
- 55. Транслокационный Синдром Патау ( = Петау = Трисомия 13).
- 56. Транслокационный Синдром Патау ( = Петау = Трисомия 13).
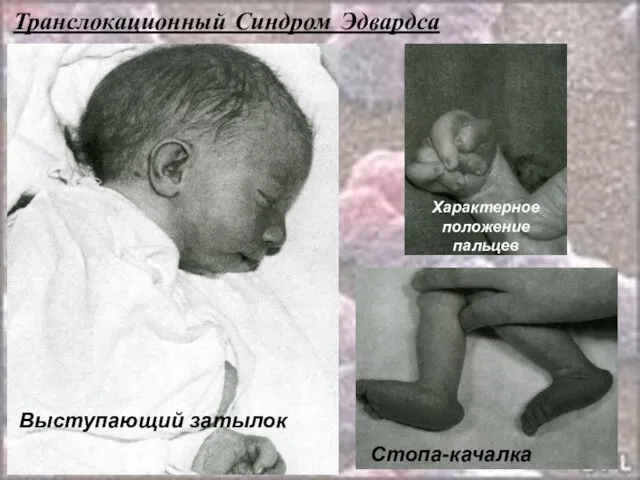
- 57. Транслокационный Синдром Эдвардса. Кариотип: 46,ХХ(ХY),E 18+/2,9,13-15,21-22 t Соотношение мальчиков и девочек равно 1:3. Причины преобладания больных
- 58. Транслокационный Синдром Эдвардса Дети с синдромом Эдвардса умирают в раннем возрасте ( 90% - до 1
- 59. Транслокационный Синдром Эдвардса Стопа-качалка Характерное положение пальцев Выступающий затылок
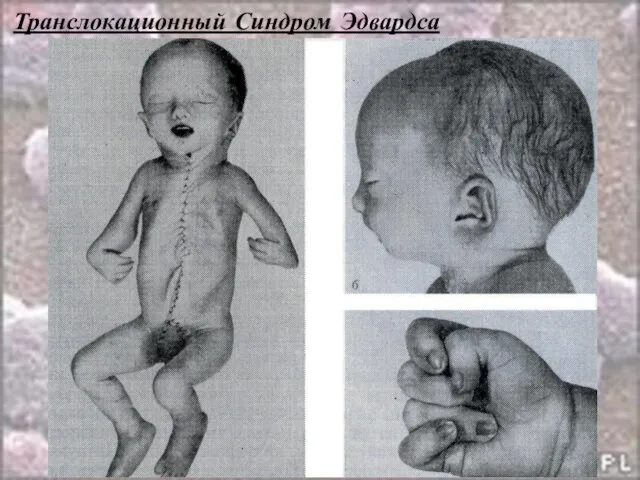
- 60. Транслокационный Синдром Эдвардса
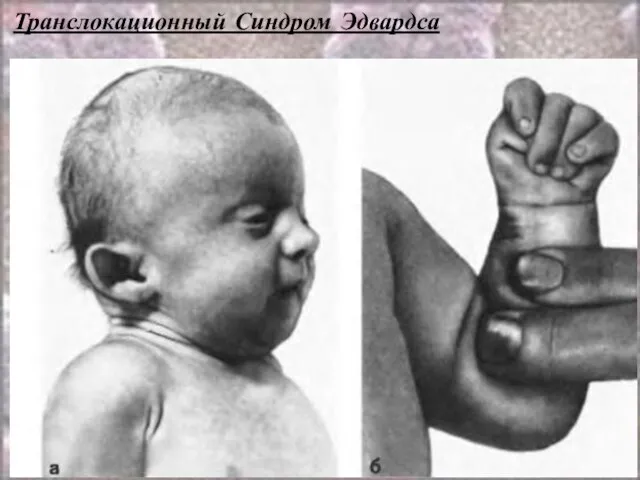
- 61. Транслокационный Синдром Эдвардса
- 64. Скачать презентацию
Слайд 2Изменчивость
Ненаследственная
(фенотипическая,
модификационная)
Наследственная
(генотипическая)
Мутационная
Комбинативная
Генные
Хромосомные
(хромосомные
аберрации)
Геномные
Полиплоидия
Гетероплоидия
Автополиплоидия
Аллополиплоидия
Внутрихромосомные
(интрахромосомные)
Межхромосомные
(интерхромосомные)
Изменчивость
Ненаследственная
(фенотипическая,
модификационная)
Наследственная
(генотипическая)
Мутационная
Комбинативная
Генные
Хромосомные
(хромосомные
аберрации)
Геномные
Полиплоидия
Гетероплоидия
Автополиплоидия
Аллополиплоидия
Внутрихромосомные
(интрахромосомные)
Межхромосомные
(интерхромосомные)
Слайд 3Мутации
Генные Хромосомные (молекулярные) болезни
болезни
Генные
Хромосомные
(хромосомные
аберрации)
Геномные
Мутации
Генные Хромосомные (молекулярные) болезни
болезни
Генные
Хромосомные
(хромосомные
аберрации)
Геномные
Слайд 4Мутации – наследуемые изменения генетического
материала, приводящие к изменению тех или иных
признаков организма.
Мутагенез
Мутации – наследуемые изменения генетического
материала, приводящие к изменению тех или иных
признаков организма.
Мутагенез
Слайд 5 Хромосомные болезни – большая группа врожденных наследственных болезней, клинически характеризующихся множественными
Хромосомные болезни – большая группа врожденных наследственных болезней, клинически характеризующихся множественными
Слайд 6 Хромосомная аномалия - обобщенное название любого типа хромосомных мутаций.
Хромосомная мутация
Хромосомная аномалия - обобщенное название любого типа хромосомных мутаций.
Хромосомная мутация
Слайд 7Основная причина возникновения различных хромосомных мутаций – разрывы хромосом и хроматид и
Основная причина возникновения различных хромосомных мутаций – разрывы хромосом и хроматид и

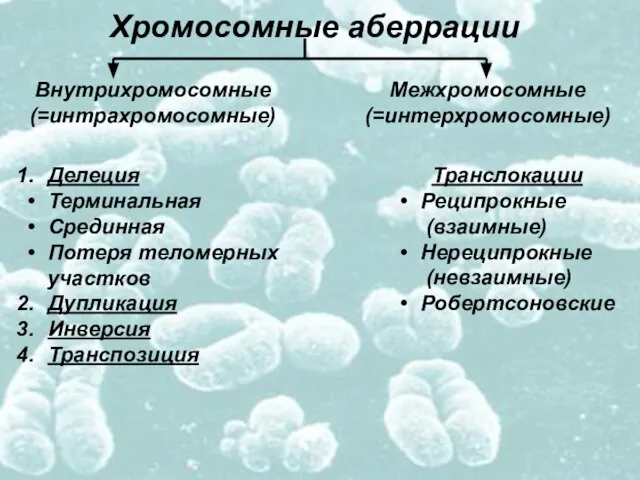
Слайд 8Хромосомные аберрации
Внутрихромосомные
(=интрахромосомные)
Межхромосомные
(=интерхромосомные)
Делеция
Терминальная
Срединная
Потеря теломерных
участков
Дупликация
Инверсия
Транспозиция
Транслокации
Реципрокные
(взаимные)
Нереципрокные
(невзаимные)
Робертсоновские
Хромосомные аберрации
Внутрихромосомные
(=интрахромосомные)
Межхромосомные
(=интерхромосомные)
Делеция
Терминальная
Срединная
Потеря теломерных
участков
Дупликация
Инверсия
Транспозиция
Транслокации
Реципрокные
(взаимные)
Нереципрокные
(невзаимные)
Робертсоновские
Слайд 91. Делеция
а) Терминальная Deficiency (=концевая)
(потеря концевых участков)
Возможна при одиночном разрыве.
1. Делеция
а) Терминальная Deficiency (=концевая)
(потеря концевых участков)
Возможна при одиночном разрыве.

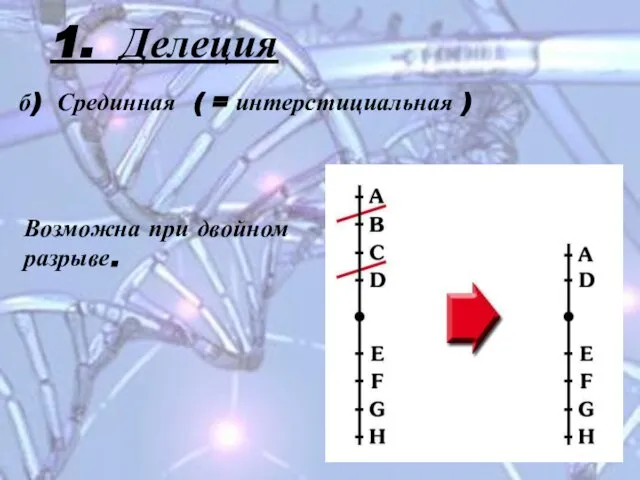
Слайд 101. Делеция
б) Срединная ( = интерстициальная )
Возможна при двойном разрыве.
1. Делеция
б) Срединная ( = интерстициальная )
Возможна при двойном разрыве.
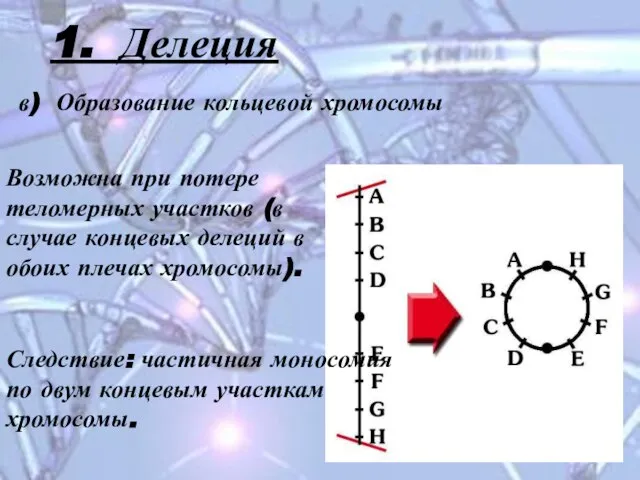
Слайд 111. Делеция
в) Образование кольцевой хромосомы
Возможна при потере теломерных участков (в случае
1. Делеция
в) Образование кольцевой хромосомы
Возможна при потере теломерных участков (в случае
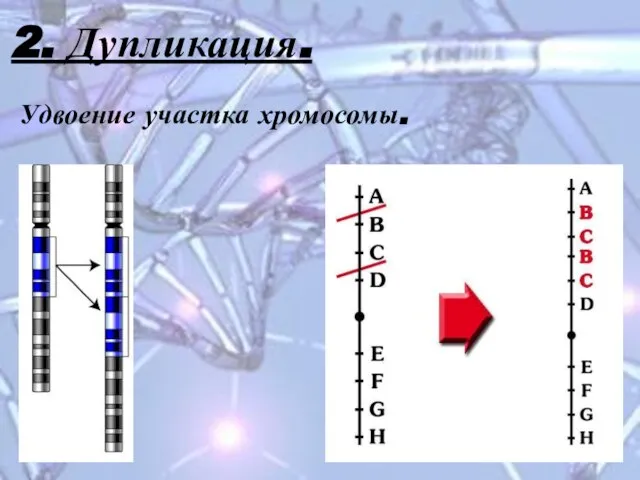
Слайд 122. Дупликация.
Удвоение участка хромосомы.
2. Дупликация.
Удвоение участка хромосомы.
Слайд 133. Инверсия
Возможна при двойном разрыве и последующем повороте образовавшегося участка на
3. Инверсия
Возможна при двойном разрыве и последующем повороте образовавшегося участка на

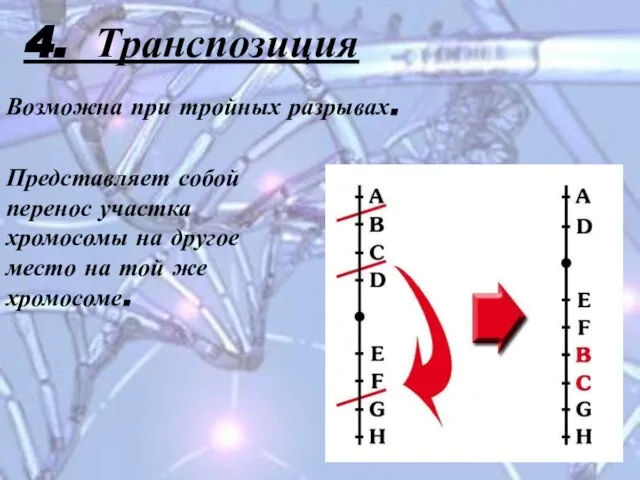
Слайд 144. Транспозиция
Возможна при тройных разрывах.
Представляет собой перенос участка хромосомы на другое
4. Транспозиция
Возможна при тройных разрывах.
Представляет собой перенос участка хромосомы на другое
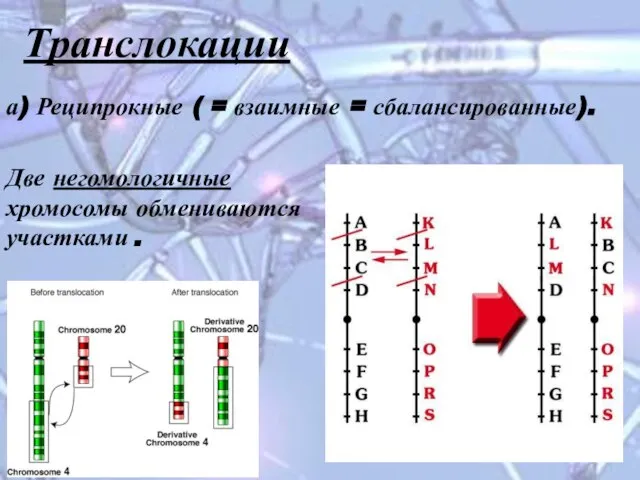
Слайд 15Транслокации
а) Реципрокные ( = взаимные = сбалансированные).
Две негомологичные хромосомы обмениваются участками
Транслокации
а) Реципрокные ( = взаимные = сбалансированные).
Две негомологичные хромосомы обмениваются участками
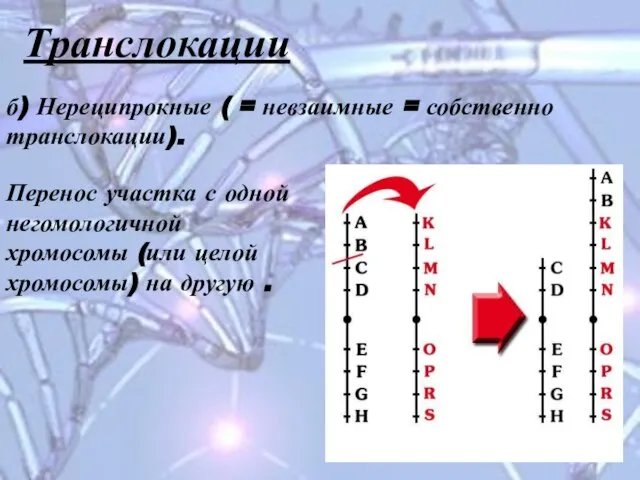
Слайд 16Транслокации
б) Нереципрокные ( = невзаимные = собственно транслокации).
Перенос участка с одной
Транслокации
б) Нереципрокные ( = невзаимные = собственно транслокации).
Перенос участка с одной
Слайд 17Транслокации
в) Робертсоновские
Две негомологичные хромосомы объединяются в одну .
(Разрыв происходит в области
Транслокации
в) Робертсоновские
Две негомологичные хромосомы объединяются в одну .
(Разрыв происходит в области

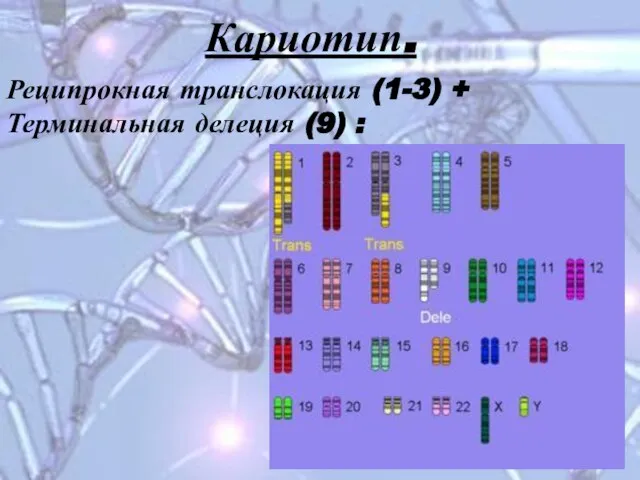
Слайд 18Реципрокная транслокация (1-3) +
Терминальная делеция (9) :
Кариотип.
Реципрокная транслокация (1-3) +
Терминальная делеция (9) :
Кариотип.
Слайд 19Робертсоновская транслокация (13;14).
Диагноз: невынашивание беременности.
Кариотип: 45,XX, rob t (13;14)
Робертсоновская транслокация (13;14).
Диагноз: невынашивание беременности.
Кариотип: 45,XX, rob t (13;14)

Слайд 20Заболевания, вызываемые
хромосомными
аберрациями
Заболевания, вызываемые
хромосомными
аберрациями
Слайд 21Синдром Лежьена
( = Синдром «Кошачьего крика» ).
Кариотип: 46,ХХ(ХY),B5p-
Частота (у новорожденных): 1:45000 –
Синдром Лежьена
( = Синдром «Кошачьего крика» ).
Кариотип: 46,ХХ(ХY),B5p-
Частота (у новорожденных): 1:45000 –

Слайд 22Синдром Лежьена
( = Синдром «Кошачьего крика» ).
Возможны и другие цитогенетические варианты:
Кольцевая хромосома
Синдром Лежьена
( = Синдром «Кошачьего крика» ).
Возможны и другие цитогенетические варианты:
Кольцевая хромосома

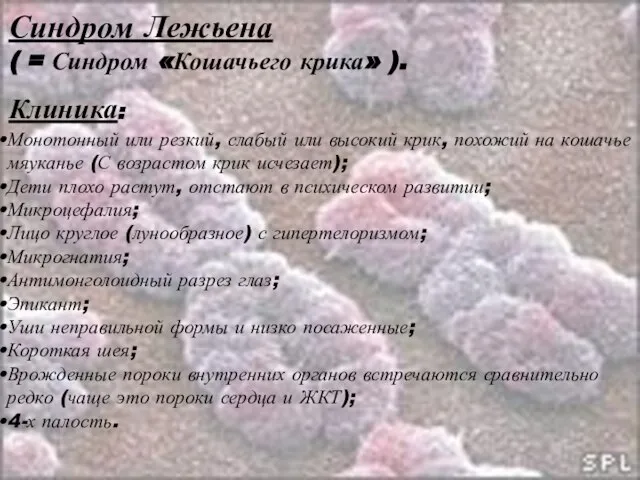
Слайд 23Синдром Лежьена
( = Синдром «Кошачьего крика» ).
Клиника:
Монотонный или резкий, слабый или высокий
Синдром Лежьена
( = Синдром «Кошачьего крика» ).
Клиника:
Монотонный или резкий, слабый или высокий
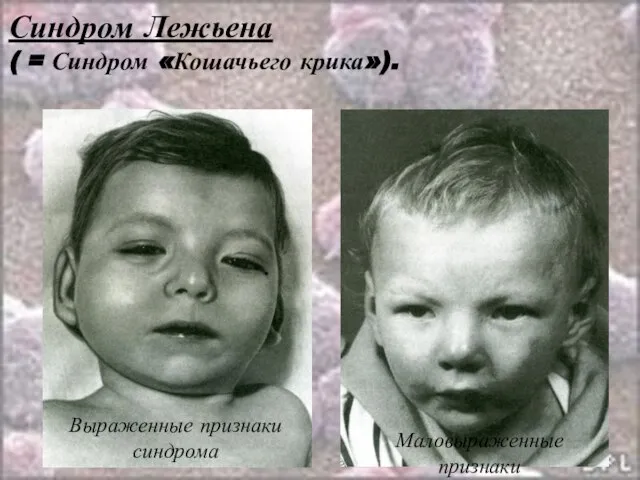
Слайд 24Синдром Лежьена
( = Синдром «Кошачьего крика»).
Выраженные признаки
синдрома
Маловыраженные
признаки
Синдром Лежьена
( = Синдром «Кошачьего крика»).
Выраженные признаки
синдрома
Маловыраженные
признаки
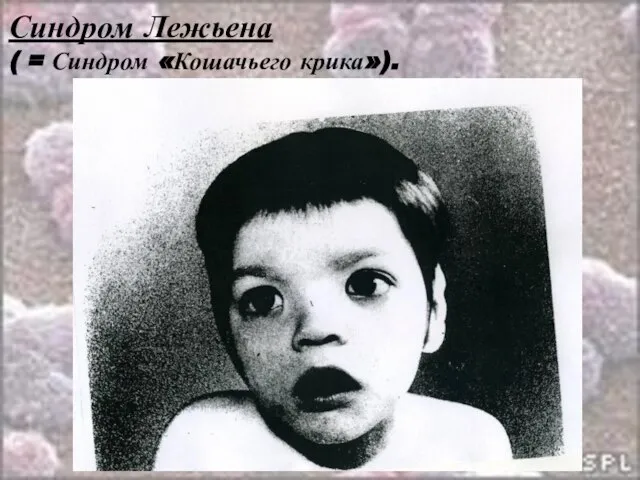
Слайд 25Синдром Лежьена
( = Синдром «Кошачьего крика»).
Синдром Лежьена
( = Синдром «Кошачьего крика»).
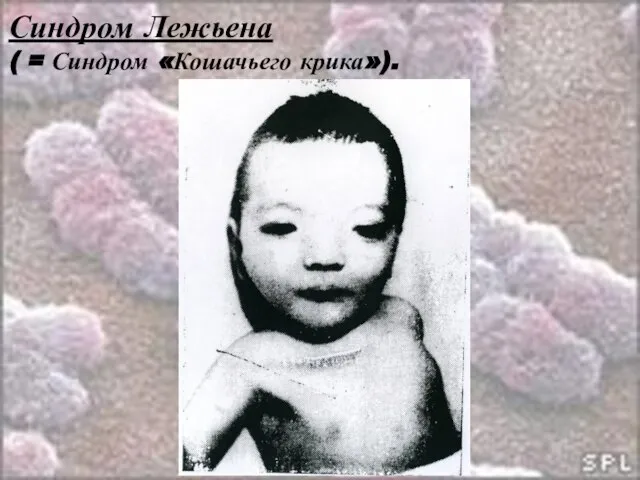
Слайд 26Синдром Лежьена
( = Синдром «Кошачьего крика»).
Синдром Лежьена
( = Синдром «Кошачьего крика»).
Слайд 27Синдром
Вольфа-Хиршхорна
Кариотип: 46,ХХ(ХY), В4р-
Обусловлен делецией короткого плеча хромосомы 4.
За симптомокомплекс ответственен сегмент 4р16
Синдром
Вольфа-Хиршхорна
Кариотип: 46,ХХ(ХY), В4р-
Обусловлен делецией короткого плеча хромосомы 4.
За симптомокомплекс ответственен сегмент 4р16

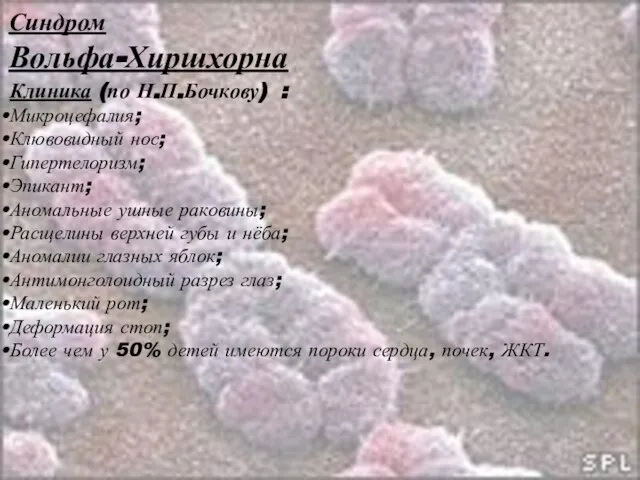
Слайд 28Синдром
Вольфа-Хиршхорна
Клиника (по Н.П.Бочкову) :
Микроцефалия;
Клювовидный нос;
Гипертелоризм;
Эпикант;
Аномальные ушные раковины;
Расщелины верхней губы и нёба;
Аномалии
Синдром
Вольфа-Хиршхорна
Клиника (по Н.П.Бочкову) :
Микроцефалия;
Клювовидный нос;
Гипертелоризм;
Эпикант;
Аномальные ушные раковины;
Расщелины верхней губы и нёба;
Аномалии
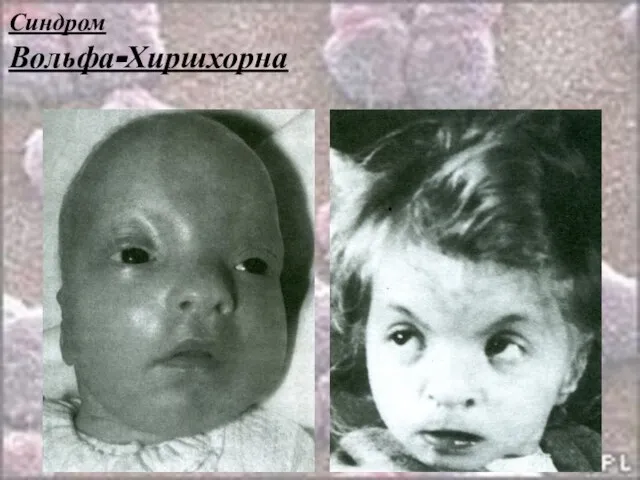
Слайд 29Синдром
Вольфа-Хиршхорна
Синдром
Вольфа-Хиршхорна
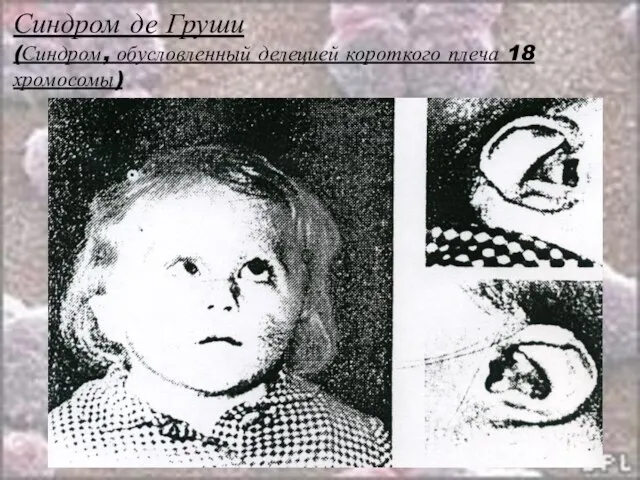
Слайд 30Синдром де Груши
(Синдром, обусловленный делецией короткого плеча 18 хромосомы)
Частота: 1:60 000
Обусловлен делецией,
Синдром де Груши
(Синдром, обусловленный делецией короткого плеча 18 хромосомы)
Частота: 1:60 000
Обусловлен делецией,

Слайд 31Синдром де Груши
(Синдром, обусловленный делецией короткого плеча 18 хромосомы)
Клиника:
Низкорослость;
Маленькая окружность черепа без
Синдром де Груши
(Синдром, обусловленный делецией короткого плеча 18 хромосомы)
Клиника:
Низкорослость;
Маленькая окружность черепа без

Слайд 32Синдром де Груши
(Синдром, обусловленный делецией короткого плеча 18 хромосомы)
Синдром де Груши
(Синдром, обусловленный делецией короткого плеча 18 хромосомы)
Слайд 33Синдром 18q-
Частота: 1:60 000
Обследовано 25 больных.
Продолжительность жизни составляет 2,5 – 5 лет.
Синдром 18q-
Частота: 1:60 000
Обследовано 25 больных.
Продолжительность жизни составляет 2,5 – 5 лет.

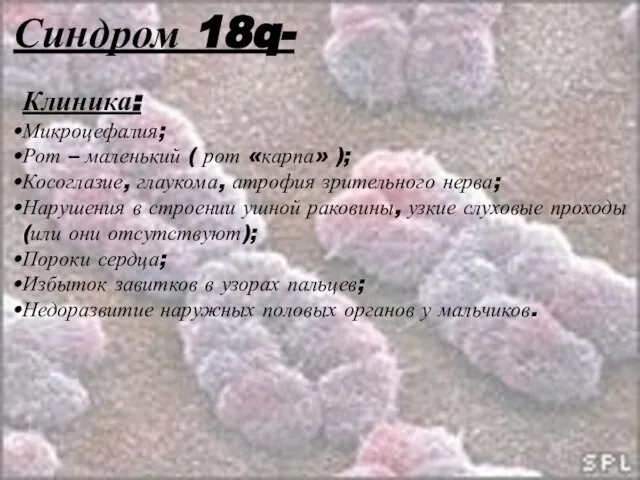
Слайд 34Синдром 18q-
Клиника:
Микроцефалия;
Рот – маленький ( рот «карпа» );
Косоглазие, глаукома, атрофия зрительного нерва;
Нарушения
Синдром 18q-
Клиника:
Микроцефалия;
Рот – маленький ( рот «карпа» );
Косоглазие, глаукома, атрофия зрительного нерва;
Нарушения
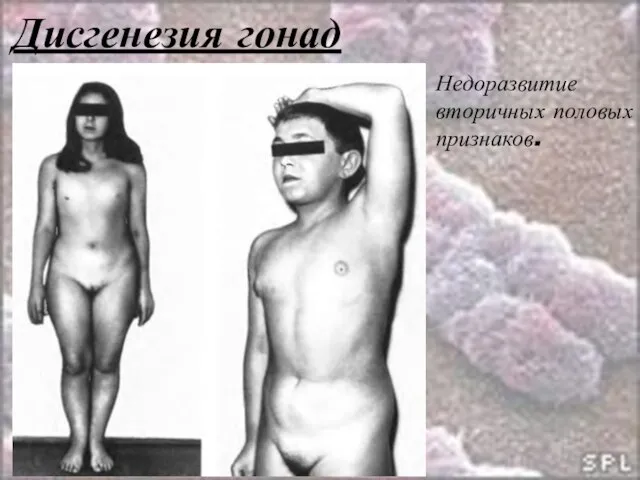
Слайд 35Дисгенезия гонад
46, ХХ р-
46, ХХ q-
Потеря плеча Х-хромосомы вызывает признаки, сходные с
Дисгенезия гонад
46, ХХ р-
46, ХХ q-
Потеря плеча Х-хромосомы вызывает признаки, сходные с

Слайд 36Дисгенезия гонад
Недоразвитие вторичных половых признаков.
Дисгенезия гонад
Недоразвитие вторичных половых признаков.
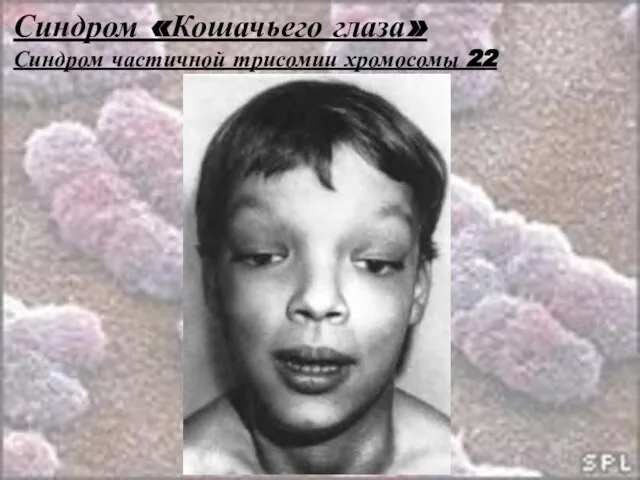
Слайд 37Синдром «Кошачьего глаза»
Синдром частичной трисомии хромосомы 22
Относится к особой группе заболеваний –
Синдром «Кошачьего глаза»
Синдром частичной трисомии хромосомы 22
Относится к особой группе заболеваний –

Слайд 38Синдром «Кошачьего глаза»
Синдром частичной трисомии хромосомы 22
Описан в 1978 году.
Встречается редко.
Частота его
Синдром «Кошачьего глаза»
Синдром частичной трисомии хромосомы 22
Описан в 1978 году.
Встречается редко.
Частота его

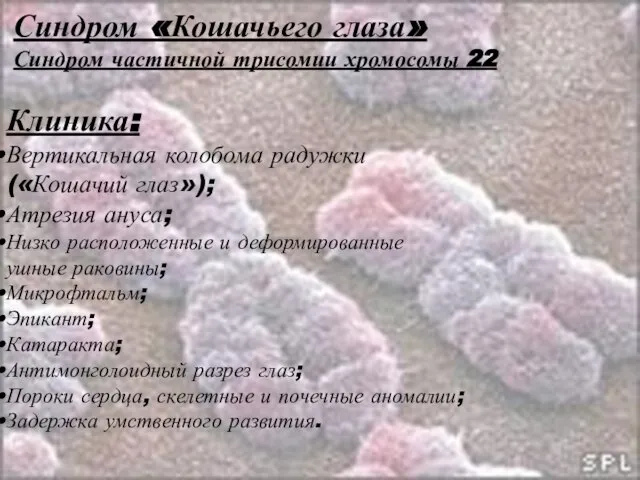
Слайд 39Синдром «Кошачьего глаза»
Синдром частичной трисомии хромосомы 22
Клиника:
Вертикальная колобома радужки
(«Кошачий глаз»);
Атрезия ануса;
Низко
Синдром «Кошачьего глаза»
Синдром частичной трисомии хромосомы 22
Клиника:
Вертикальная колобома радужки
(«Кошачий глаз»);
Атрезия ануса;
Низко
Слайд 40Синдром «Кошачьего глаза»
Синдром частичной трисомии хромосомы 22
Синдром «Кошачьего глаза»
Синдром частичной трисомии хромосомы 22
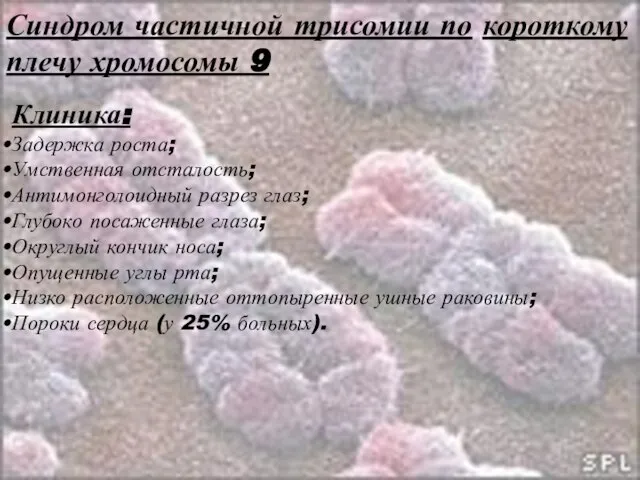
Слайд 41Синдром частичной трисомии по короткому плечу хромосомы 9
Обследовано более 200 больных.
Является результатом
Синдром частичной трисомии по короткому плечу хромосомы 9
Обследовано более 200 больных.
Является результатом

Слайд 42Синдром частичной трисомии по короткому плечу хромосомы 9
Клиника:
Задержка роста;
Умственная отсталость;
Антимонголоидный разрез глаз;
Глубоко
Синдром частичной трисомии по короткому плечу хромосомы 9
Клиника:
Задержка роста;
Умственная отсталость;
Антимонголоидный разрез глаз;
Глубоко
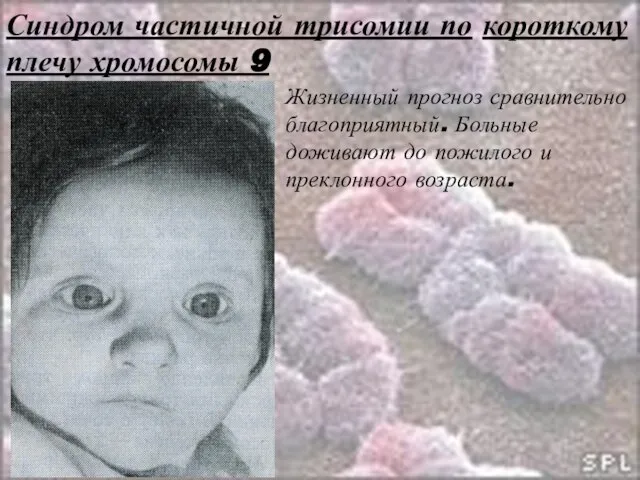
Слайд 43Синдром частичной трисомии по короткому плечу хромосомы 9
Жизненный прогноз сравнительно благоприятный. Больные
Синдром частичной трисомии по короткому плечу хромосомы 9
Жизненный прогноз сравнительно благоприятный. Больные
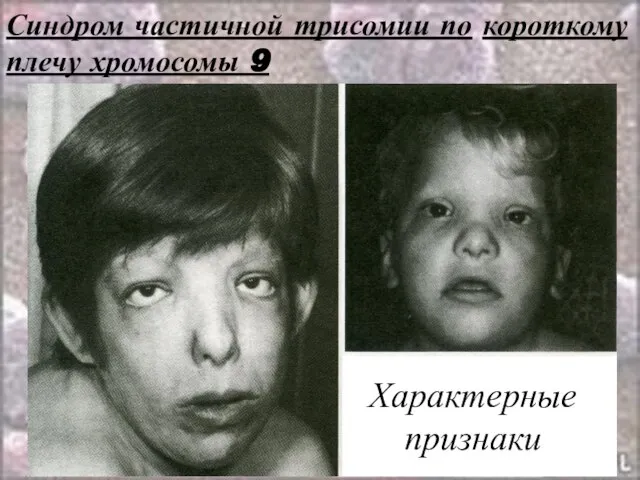
Слайд 44Синдром частичной трисомии по короткому плечу хромосомы 9
Характерные
признаки
Синдром частичной трисомии по короткому плечу хромосомы 9
Характерные
признаки
Слайд 45Синдром Орбели
(частичной моносомии 13q-)
Частота 1:100 000
Обусловлен образованием на длинном плече хромосомы 13
Синдром Орбели
(частичной моносомии 13q-)
Частота 1:100 000
Обусловлен образованием на длинном плече хромосомы 13

Слайд 46Синдром Орбели
(частичной моносомии 13q-)
Клиника:
Лицо асимметричное
Широкая, выступающая спинка носа – «Греческий профиль»
Эпикант
Микрофтальмия
Колобома радужки
Синдром Орбели
(частичной моносомии 13q-)
Клиника:
Лицо асимметричное
Широкая, выступающая спинка носа – «Греческий профиль»
Эпикант
Микрофтальмия
Колобома радужки

Слайд 47Хронический миелолейкоз
У 95% больных в опухолевых клетках обнаруживается укороченная 22-я «Филадельфийская» хромосома,
Хронический миелолейкоз
У 95% больных в опухолевых клетках обнаруживается укороченная 22-я «Филадельфийская» хромосома,

Слайд 48Хронический миелолейкоз
Отмечаются:
Изменение лейкоцитарной формулы (сдвиг формулы влево)
Выраженная эозинофилия и базофилия
Анемия
Наличие множества ядросодержащих
Хронический миелолейкоз
Отмечаются:
Изменение лейкоцитарной формулы (сдвиг формулы влево)
Выраженная эозинофилия и базофилия
Анемия
Наличие множества ядросодержащих
Слайд 49Транслокационный
Синдром Дауна
46,ХХ(ХY),G21+/2,9,13-15,21-22t
Примерно 3-4% больных с синдромом Дауна имеют транслокационную форму трисомии по
Транслокационный
Синдром Дауна
46,ХХ(ХY),G21+/2,9,13-15,21-22t
Примерно 3-4% больных с синдромом Дауна имеют транслокационную форму трисомии по

Слайд 50Транслокационный
Синдром Дауна
Клиника:
Микроцефалия
Умственная отсталость
Голова уменьшенных размеров, череп круглый, затылок плоский
Лицо плоское, нос короткий
Транслокационный
Синдром Дауна
Клиника:
Микроцефалия
Умственная отсталость
Голова уменьшенных размеров, череп круглый, затылок плоский
Лицо плоское, нос короткий

Слайд 51Транслокационный
Синдром Дауна
Транслокационный
Синдром Дауна
Слайд 52Транслокационный
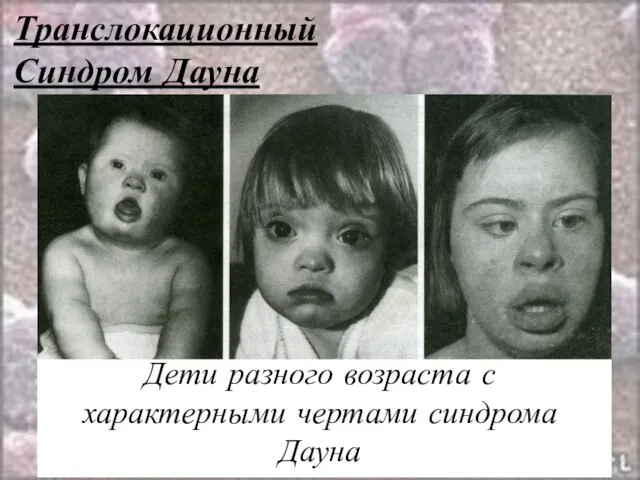
Синдром Дауна
Дети разного возраста с характерными чертами синдрома Дауна
Транслокационный
Синдром Дауна
Дети разного возраста с характерными чертами синдрома Дауна
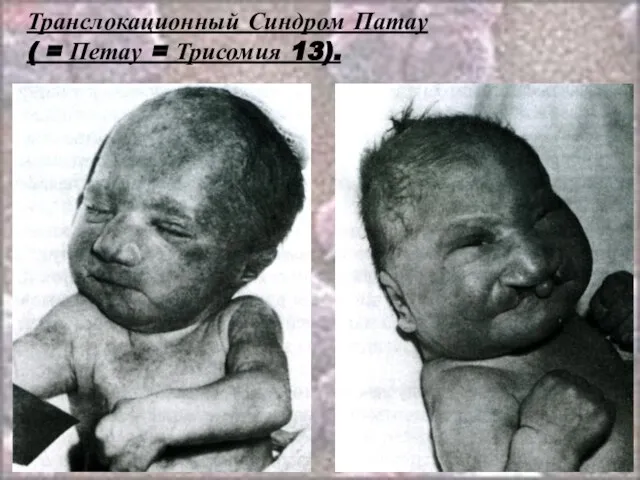
Слайд 53Транслокационный Синдром Патау
( = Петау = Трисомия 13).
Кариотип: 46,ХХ(ХY),D 13+/2,9,13-15,21-22 t
Соотношение полов
Транслокационный Синдром Патау
( = Петау = Трисомия 13).
Кариотип: 46,ХХ(ХY),D 13+/2,9,13-15,21-22 t
Соотношение полов

Слайд 54Транслокационный Синдром Патау
( = Петау = Трисомия 13).
В связи с тяжелыми
Транслокационный Синдром Патау
( = Петау = Трисомия 13).
В связи с тяжелыми

Слайд 55Транслокационный Синдром Патау
( = Петау = Трисомия 13).
Транслокационный Синдром Патау
( = Петау = Трисомия 13).
Слайд 56Транслокационный Синдром Патау
( = Петау = Трисомия 13).
Транслокационный Синдром Патау
( = Петау = Трисомия 13).
Слайд 57Транслокационный
Синдром Эдвардса.
Кариотип: 46,ХХ(ХY),E 18+/2,9,13-15,21-22 t
Соотношение мальчиков и девочек равно 1:3. Причины преобладания
Транслокационный
Синдром Эдвардса.
Кариотип: 46,ХХ(ХY),E 18+/2,9,13-15,21-22 t
Соотношение мальчиков и девочек равно 1:3. Причины преобладания

Слайд 58Транслокационный Синдром Эдвардса
Дети с синдромом Эдвардса умирают в раннем возрасте (
Транслокационный Синдром Эдвардса
Дети с синдромом Эдвардса умирают в раннем возрасте (

Слайд 59Транслокационный Синдром Эдвардса
Стопа-качалка
Характерное
положение
пальцев
Выступающий затылок
Транслокационный Синдром Эдвардса
Стопа-качалка
Характерное
положение
пальцев
Выступающий затылок
Слайд 60Транслокационный Синдром Эдвардса
Транслокационный Синдром Эдвардса
Слайд 61Транслокационный Синдром Эдвардса
Транслокационный Синдром Эдвардса

 Фитнес-индустрия в Архангельске.
Фитнес-индустрия в Архангельске. РЕСПУБЛИКАНСКИЙ СЕМИНАР-СОВЕЩАНИЕ НА ТЕМУ: «ОРГАНИЗАЦИЯ ДЕЯТЕЛЬНОСТИ ПО ПЕРЕВОЗКЕ ПАССАЖИРОВ И БАГАЖА ЛЕГКОВЫМ ТАКСИ»
РЕСПУБЛИКАНСКИЙ СЕМИНАР-СОВЕЩАНИЕ НА ТЕМУ: «ОРГАНИЗАЦИЯ ДЕЯТЕЛЬНОСТИ ПО ПЕРЕВОЗКЕ ПАССАЖИРОВ И БАГАЖА ЛЕГКОВЫМ ТАКСИ» Нетрадиционный урок истории
Нетрадиционный урок истории www.sales.ua
www.sales.ua Презентация на тему Речь (8 класс)
Презентация на тему Речь (8 класс) Выбор доменного имени
Выбор доменного имени  Акробатические элементы. Упражнения на гибкость, растяжка, координация
Акробатические элементы. Упражнения на гибкость, растяжка, координация Присоединение Прибалтики к СССР
Присоединение Прибалтики к СССР Таможенное оформление
Таможенное оформление Соединения азота
Соединения азота Образовательные запросы родителей
Образовательные запросы родителей Прогулки по Самаре
Прогулки по Самаре Зарождение
Зарождение Базы данных
Базы данных Комментарии по доработке макетов батончиков Racionika Protein
Комментарии по доработке макетов батончиков Racionika Protein Прямоугольное проецирование на три плоскости проекций
Прямоугольное проецирование на три плоскости проекций Мой посёлок – моя гордость
Мой посёлок – моя гордость Цинк для КРС в премиксах и функциональных кормовых продуктах
Цинк для КРС в премиксах и функциональных кормовых продуктах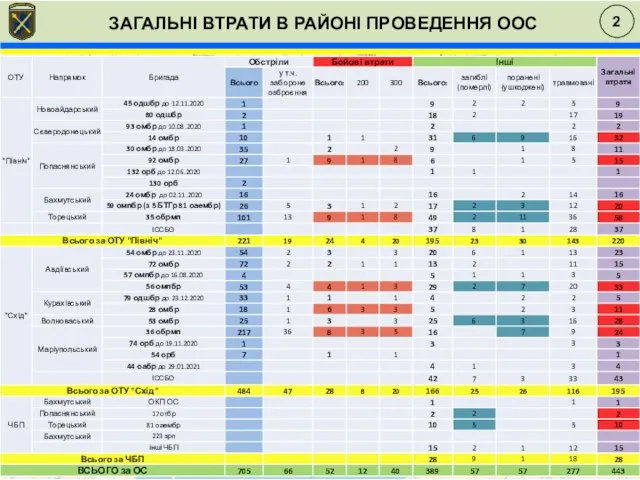 Загальні втрати в районі проведення ООС
Загальні втрати в районі проведення ООС «Либэр. Электронная библиотека» - новый этап в автоматизации Программное решение для создания электронной библиотеки
«Либэр. Электронная библиотека» - новый этап в автоматизации Программное решение для создания электронной библиотеки Поздравление с Новым годом
Поздравление с Новым годом Свойства эфирных масел по степени воздействия на ЦНС
Свойства эфирных масел по степени воздействия на ЦНС Презентация на тему Древние люди: где искать наши корни
Презентация на тему Древние люди: где искать наши корни Эволюция телефона за 100 лет
Эволюция телефона за 100 лет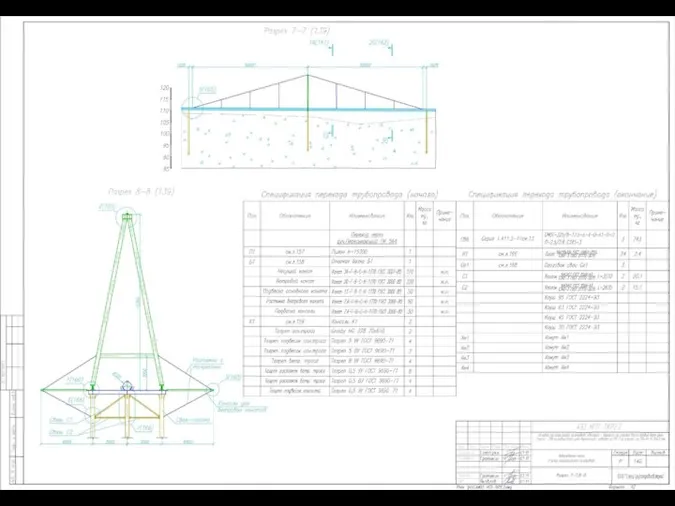 Противопучинные мероприятия сваи СМОТ-325 газопровод
Противопучинные мероприятия сваи СМОТ-325 газопровод CMC Kazakhstan и Консорциум Консалтинговых и исследовательских компаний Казахстана
CMC Kazakhstan и Консорциум Консалтинговых и исследовательских компаний Казахстана Количественный анализ аскорбиновой кислоты
Количественный анализ аскорбиновой кислоты Kunstarten
Kunstarten