- Обработка данных секвенирования

Содержание
- 2. Покрытие Покрытие (глубина секвенирования) – важный параметр методов NGS: кратность прочтения каждого нуклеотида. Для каждой задачи
- 3. Оценка необходимого покрытия Вероятность того, что нуклеотид не будет определён (P), исходя из глубины покрытия (c)
- 4. Анализ данных секвенирования 1. Очистка “сырых” данных (raw data) (фильтрация ридов по качеству). Результат: “примесные” риды
- 5. 1. Оценка качества ридов: FASTQ – формат записи ридов @SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65 Каждая последовательность занимает
- 6. Определение качества ридов по шкале Phred Каждый символ означает какое-то число (Q) от 0 до 100.
- 7. Phred и ASCII: номера присваивают начиная с 33 символа (!=0) (Phred+33) или с 64 (@=0) (Phred+64)
- 8. Кодировка качества Phred+33
- 9. Примеры качества по шкалам Phred+33 и Phred+64
- 10. Источники ошибок в ридах: примеси Примеси бывают: 1. Артефактные (ошибки секвенирования) образование димеров адаптеров чтение сквозь
- 11. Источники ошибок в ридах: фазировка Фрагменты в одном кластере строятся с разной скоростью – секвенатору сложно
- 12. Программа FastQC – контроль качества ридов: 1. Среднее нуклеотидное качество – хорошее (все Me>25, все Q1>10)
- 13. Программа FastQC – контроль качества ридов: 1. Среднее нуклеотидное качество – неудовлетворительное (есть Me
- 14. Программа FastQC – контроль качества ридов: 2. Средний нуклеотидный состав ридов
- 15. Программа FastQC – контроль качества ридов: 2. Средний нуклеотидный состав ридов
- 16. Программа FastQC – контроль качества ридов: 3. Чрезмерно представленные последовательности
- 17. Очистка “сырых” ридов: тримминг 1. Удаление адаптерных последовательностей из ридов 2. Отсечение с конца ридов нуклеотидов,
- 18. Особый этап для метагеномики – Сортировка данных (биннинг) 1. Методы, основанные на нуклеотидном составе GC-состав динуклеотидный
- 19. 2. Сборка генома (assemby) de novo (сборка не секвенированного ранее генома) – метод OLC (overlap layout
- 20. Сборка de novo: Overlap layout consensus: 1 Поиск пар ридов, имеющих общие k-меры (последовательности длиной k,
- 21. Сборка de novo: Overlap layout consensus: 2 На базе попарного выравнивания строят множественное выравнивание, корректируют ошибки
- 22. Сборка de novo: Графы де Брёйна
- 23. Результат сборки: контиги и скэффолды
- 24. Качество сборки генома N50 – длина контига, который вместе с остальными контигами большей длины покрывает не
- 25. Формат представления нуклеотидных последовательностей – FASTA >OTU-160-1 Acinetobacter baumannii CCTACGGGGGGCTGCAGTGGGGAATATTGGACAATGGGGGGAACCCTGATCCAGCCATGCCGCGTGTGTGAAGAAGGCCTTATGGTTGTAAAGCACTTTAAGCGAGGAGGAGGCTACTCTAGTTAATACCTAGGGATAGTGGACGTTACTCGCAGAATAA Каждая последовательность занимает две строки: 1).
- 26. 3. Аннотация 1. Поиск белок-кодирующих последовательностей на основе гомологии – сравнение с уже известными генами аннотация
- 28. Скачать презентацию
Слайд 2Покрытие
Покрытие (глубина секвенирования) – важный параметр методов NGS: кратность прочтения каждого нуклеотида.
Покрытие
Покрытие (глубина секвенирования) – важный параметр методов NGS: кратность прочтения каждого нуклеотида.

Слайд 3Оценка необходимого покрытия
Вероятность того, что нуклеотид не будет определён (P), исходя из
Оценка необходимого покрытия
Вероятность того, что нуклеотид не будет определён (P), исходя из

Слайд 4Анализ данных секвенирования
1. Очистка “сырых” данных (raw data) (фильтрация ридов по качеству).
Результат:
Анализ данных секвенирования
1. Очистка “сырых” данных (raw data) (фильтрация ридов по качеству).
Результат:

Слайд 51. Оценка качества ридов:
FASTQ – формат записи ридов
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
Каждая последовательность занимает 4
1. Оценка качества ридов:
FASTQ – формат записи ридов
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
Каждая последовательность занимает 4

Слайд 6Определение качества ридов по шкале Phred
Каждый символ означает какое-то число (Q) от
Определение качества ридов по шкале Phred
Каждый символ означает какое-то число (Q) от

Слайд 7Phred и ASCII:
номера присваивают начиная с 33 символа (!=0) (Phred+33) или с
Phred и ASCII: номера присваивают начиная с 33 символа (!=0) (Phred+33) или с
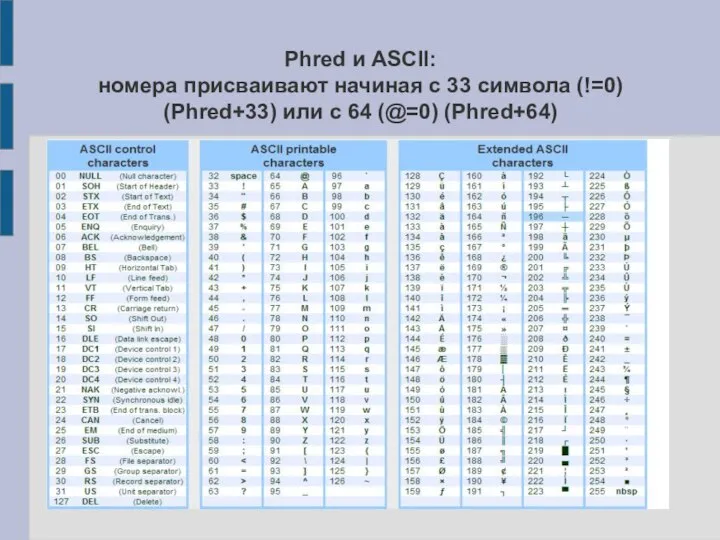
Слайд 8Кодировка качества Phred+33
Кодировка качества Phred+33

Слайд 9Примеры качества по шкалам Phred+33 и Phred+64
Примеры качества по шкалам Phred+33 и Phred+64
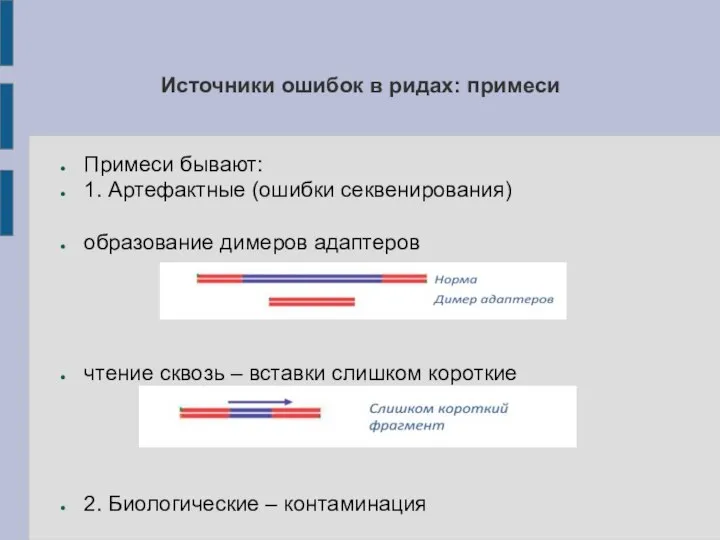
Слайд 10Источники ошибок в ридах: примеси
Примеси бывают:
1. Артефактные (ошибки секвенирования)
образование димеров адаптеров
чтение сквозь
Источники ошибок в ридах: примеси
Примеси бывают:
1. Артефактные (ошибки секвенирования)
образование димеров адаптеров
чтение сквозь
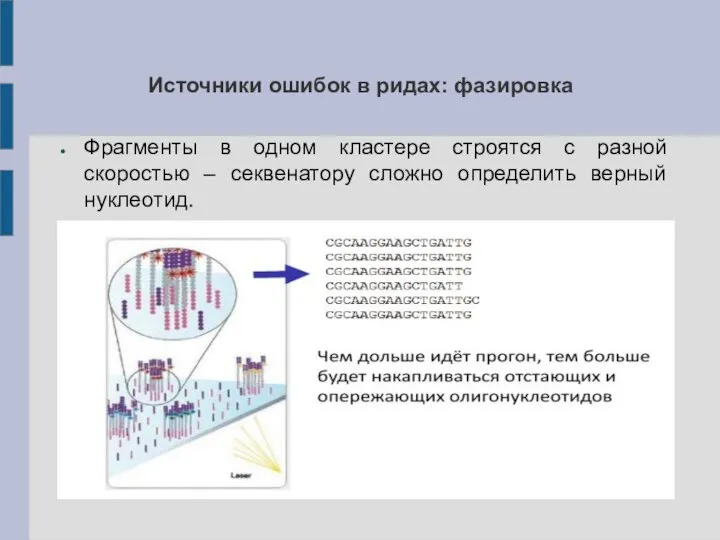
Слайд 11Источники ошибок в ридах: фазировка
Фрагменты в одном кластере строятся с разной скоростью
Источники ошибок в ридах: фазировка
Фрагменты в одном кластере строятся с разной скоростью
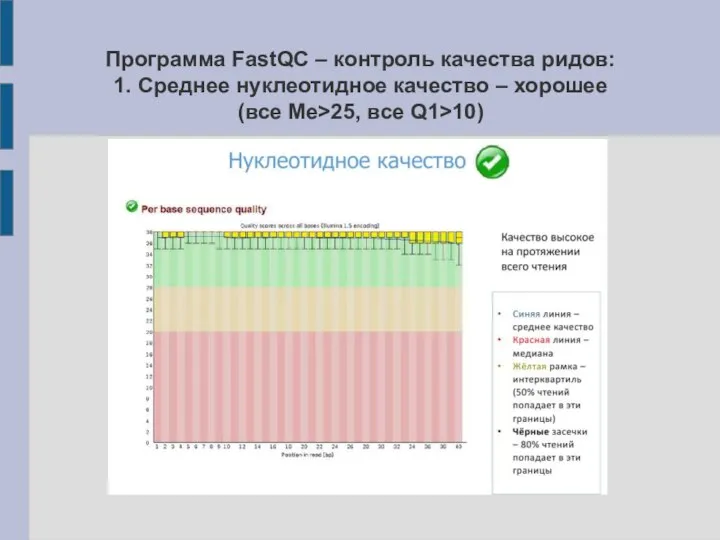
Слайд 12Программа FastQC – контроль качества ридов:
1. Среднее нуклеотидное качество – хорошее
(все Me>25,
Программа FastQC – контроль качества ридов: 1. Среднее нуклеотидное качество – хорошее (все Me>25,
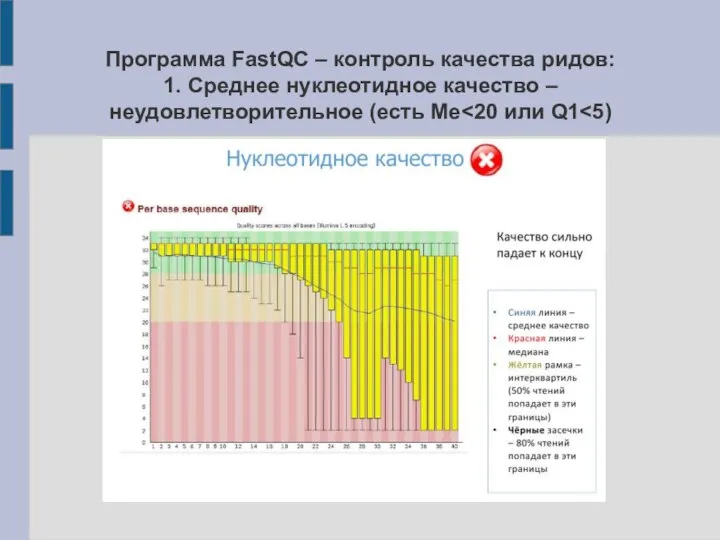
Слайд 13Программа FastQC – контроль качества ридов:
1. Среднее нуклеотидное качество – неудовлетворительное (есть
Программа FastQC – контроль качества ридов: 1. Среднее нуклеотидное качество – неудовлетворительное (есть
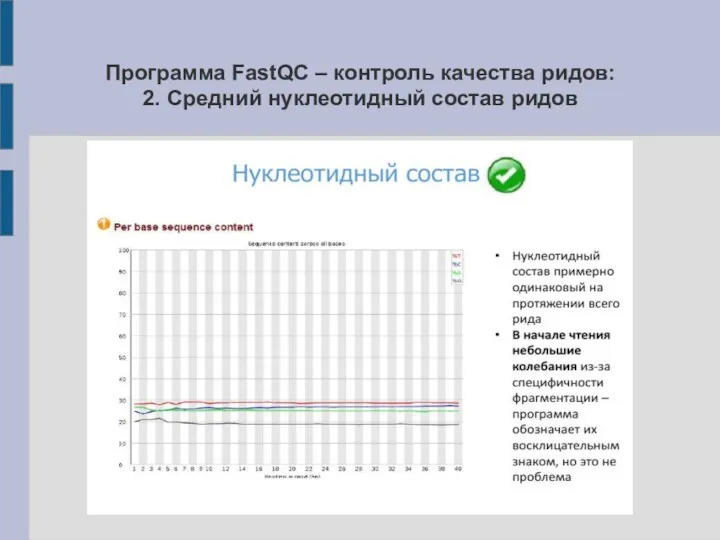
Слайд 14Программа FastQC – контроль качества ридов:
2. Средний нуклеотидный состав ридов
Программа FastQC – контроль качества ридов:
2. Средний нуклеотидный состав ридов
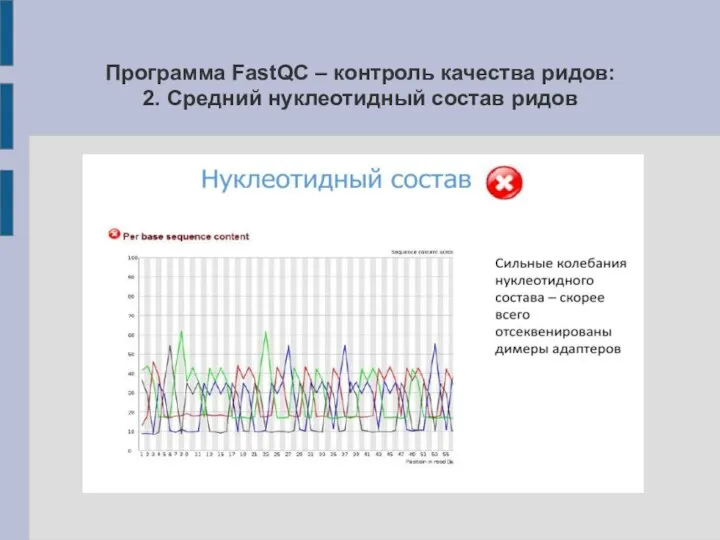
Слайд 15Программа FastQC – контроль качества ридов:
2. Средний нуклеотидный состав ридов
Программа FastQC – контроль качества ридов:
2. Средний нуклеотидный состав ридов
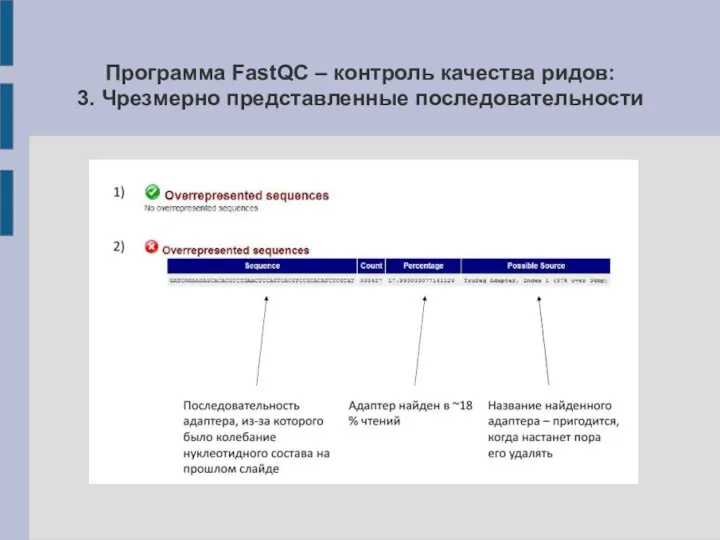
Слайд 16Программа FastQC – контроль качества ридов:
3. Чрезмерно представленные последовательности
Программа FastQC – контроль качества ридов:
3. Чрезмерно представленные последовательности
Слайд 17Очистка “сырых” ридов: тримминг
1. Удаление адаптерных последовательностей из ридов
2. Отсечение с конца
Очистка “сырых” ридов: тримминг
1. Удаление адаптерных последовательностей из ридов
2. Отсечение с конца

Слайд 18Особый этап для метагеномики – Сортировка данных (биннинг)
1. Методы, основанные на нуклеотидном
Особый этап для метагеномики – Сортировка данных (биннинг)
1. Методы, основанные на нуклеотидном

Слайд 192. Сборка генома (assemby)
de novo (сборка не секвенированного ранее генома)
– метод OLC
2. Сборка генома (assemby)
de novo (сборка не секвенированного ранее генома)
– метод OLC

Слайд 20Сборка de novo: Overlap layout consensus: 1
Поиск пар ридов, имеющих общие k-меры
Сборка de novo: Overlap layout consensus: 1
Поиск пар ридов, имеющих общие k-меры

Слайд 21Сборка de novo: Overlap layout consensus: 2
На базе попарного выравнивания строят множественное
Сборка de novo: Overlap layout consensus: 2
На базе попарного выравнивания строят множественное

Слайд 22Сборка de novo: Графы де Брёйна
Сборка de novo: Графы де Брёйна

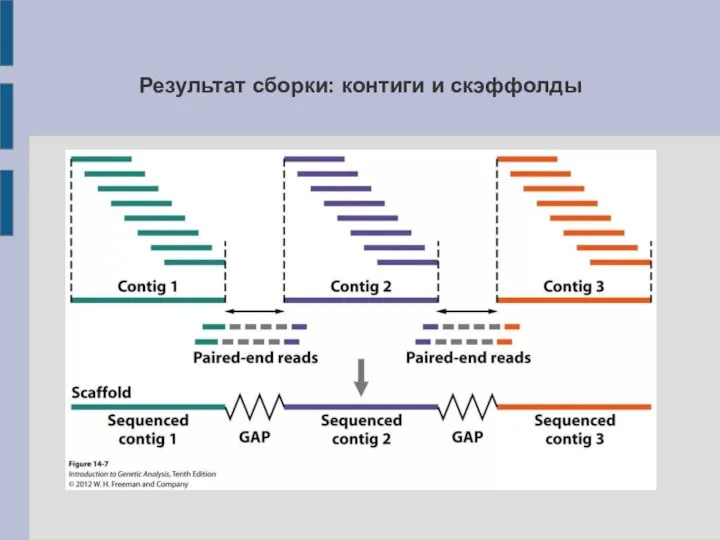
Слайд 23Результат сборки: контиги и скэффолды
Результат сборки: контиги и скэффолды
Слайд 24Качество сборки генома
N50 – длина контига, который вместе с остальными контигами большей
Качество сборки генома
N50 – длина контига, который вместе с остальными контигами большей

Слайд 25Формат представления нуклеотидных последовательностей – FASTA
>OTU-160-1 Acinetobacter baumannii
CCTACGGGGGGCTGCAGTGGGGAATATTGGACAATGGGGGGAACCCTGATCCAGCCATGCCGCGTGTGTGAAGAAGGCCTTATGGTTGTAAAGCACTTTAAGCGAGGAGGAGGCTACTCTAGTTAATACCTAGGGATAGTGGACGTTACTCGCAGAATAA
Каждая последовательность занимает две строки:
1).
Формат представления нуклеотидных последовательностей – FASTA
>OTU-160-1 Acinetobacter baumannii
CCTACGGGGGGCTGCAGTGGGGAATATTGGACAATGGGGGGAACCCTGATCCAGCCATGCCGCGTGTGTGAAGAAGGCCTTATGGTTGTAAAGCACTTTAAGCGAGGAGGAGGCTACTCTAGTTAATACCTAGGGATAGTGGACGTTACTCGCAGAATAA
Каждая последовательность занимает две строки:
1).

Слайд 263. Аннотация
1. Поиск белок-кодирующих последовательностей
на основе гомологии – сравнение с уже известными
3. Аннотация
1. Поиск белок-кодирующих последовательностей
на основе гомологии – сравнение с уже известными

 Брюхоногие моллюски
Брюхоногие моллюски Построение вариационной кривой
Построение вариационной кривой Презентация на тему Особенности скелета человека
Презентация на тему Особенности скелета человека  PPT Energy-1
PPT Energy-1 Хвойные растения
Хвойные растения According to the soil specialization. (And some botanical science)
According to the soil specialization. (And some botanical science) Интересная зоология: разработка комплекса приёмов работы с детьми по обобщению школьного курса зоологии в 7 классе
Интересная зоология: разработка комплекса приёмов работы с детьми по обобщению школьного курса зоологии в 7 классе Подземный житель — крот
Подземный житель — крот Приспособленность растений к жизни в дубраве
Приспособленность растений к жизни в дубраве Презентация на тему Мой родной край виртуальное путешествие
Презентация на тему Мой родной край виртуальное путешествие  Как растет чай
Как растет чай Класс Пресмыкающиеся. Отряд Чешуйчатые
Класс Пресмыкающиеся. Отряд Чешуйчатые Презентация на тему ПРЕДМЕТ И ЗАДАЧИ БИОТЕХНОЛОГИИ
Презентация на тему ПРЕДМЕТ И ЗАДАЧИ БИОТЕХНОЛОГИИ  Құрғақ және қуаң дала фаунасы
Құрғақ және қуаң дала фаунасы Системы объектов
Системы объектов Презентация на тему ИСПАРЕНИЕ ВОДЫ РАСТЕНИЯМИ
Презентация на тему ИСПАРЕНИЕ ВОДЫ РАСТЕНИЯМИ  Свет и зрение
Свет и зрение Естественные защитные силы организма
Естественные защитные силы организма Опыление и оплодотворение у растений
Опыление и оплодотворение у растений Плоские черви. Урок 3
Плоские черви. Урок 3 Вегетативная нервная система
Вегетативная нервная система Пчеловодство
Пчеловодство Законы генетики. Врожденная близорукость
Законы генетики. Врожденная близорукость Законы наследственности. Типы гибридизации
Законы наследственности. Типы гибридизации Покрытосеменные растения
Покрытосеменные растения Сходство зародышей и эмбриональная дивергенция. Биогенетический закон. Развитие организмов и окружающая среда
Сходство зародышей и эмбриональная дивергенция. Биогенетический закон. Развитие организмов и окружающая среда Возр. особенности скелета (2)
Возр. особенности скелета (2) Лекарственные растения и их роль в жизни человека
Лекарственные растения и их роль в жизни человека