- HyperChem HOW to USE

Содержание
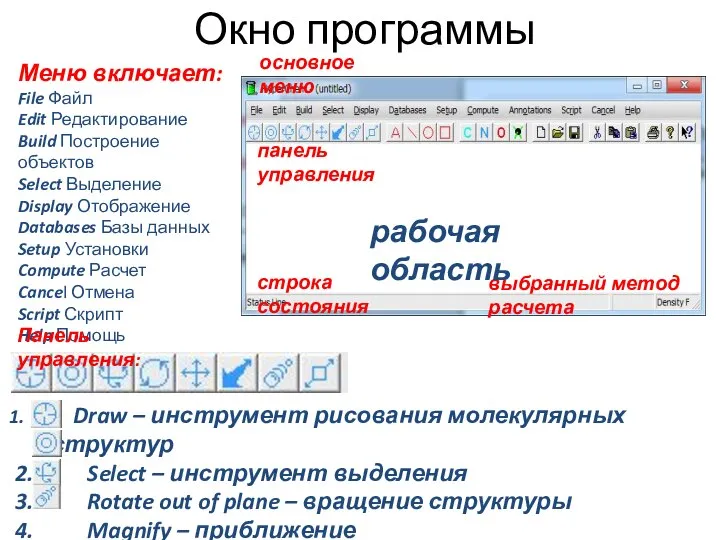
- 2. Окно программы Меню включает: File Файл Edit Редактирование Build Построение объектов Select Выделение Display Отображение Databases
- 3. Рисование молекулярных структур 1. Откройте в меню Build пункт Default element (периодическая таблица элементов Д. И.
- 4. Работа с меню Select: 1) Atoms – выделение отдельных атомов 2) Molecules – выделение молекул целиком
- 5. Измерение длины связи – необходимо выделить связь, в строке состояния появляется значение длины связи между двумя
- 6. Создание и редактирование молекулярных структур в 2D и 3D При построении чаще всего получается неверная начальная
- 7. Маркировка атомов: В меню Display нужно выбрать Labels (Этикетки) Можно выбрать Symbol, Name, Number и пр.
- 8. Расчеты в HyperChem (меню Setup)
- 10. Методы молекулярной механики
- 11. Диалоговое окно полуэмпирического метода Total charge (Полный заряд системы) вычисляется как разность между полным количеством электронов
- 12. Convergence Limit (Параметр сходимости). SCF расчет заканчивается тогда, когда отличия в полной энергии двух последующих итераций
- 13. Ab initio (неэмпирические методы расчета) Базисный набор (basic set) – это набор функций, используемых для аппроксимации
- 15. Скачать презентацию
Слайд 2Окно программы
Меню включает:
File Файл
Edit Редактирование
Build Построение объектов
Select Выделение
Display Отображение
Databases Базы данных
Setup Установки
Compute Расчет
Cancel Отмена
Script Скрипт
Help Помощь
Панель управления:
Окно программы
Меню включает:
File Файл
Edit Редактирование
Build Построение объектов
Select Выделение
Display Отображение
Databases Базы данных
Setup Установки
Compute Расчет
Cancel Отмена
Script Скрипт
Help Помощь
Панель управления:
Слайд 3Рисование молекулярных структур
1. Откройте в меню Build пункт Default element (периодическая таблица
Рисование молекулярных структур
1. Откройте в меню Build пункт Default element (периодическая таблица

Слайд 4Работа с меню Select:
1) Atoms – выделение отдельных атомов
2) Molecules – выделение
Работа с меню Select:
1) Atoms – выделение отдельных атомов
2) Molecules – выделение

Слайд 5Измерение длины связи – необходимо выделить связь, в строке состояния появляется значение
Измерение длины связи – необходимо выделить связь, в строке состояния появляется значение

Слайд 6Создание и редактирование молекулярных структур в 2D и 3D
При построении чаще всего
Создание и редактирование молекулярных структур в 2D и 3D
При построении чаще всего

Слайд 7Маркировка атомов:
В меню Display нужно выбрать Labels (Этикетки)
Можно выбрать Symbol, Name, Number
Маркировка атомов:
В меню Display нужно выбрать Labels (Этикетки)
Можно выбрать Symbol, Name, Number

Слайд 8Расчеты в HyperChem
(меню Setup)
Расчеты в HyperChem
(меню Setup)


Слайд 10Методы молекулярной механики
Методы молекулярной механики

Слайд 11Диалоговое окно полуэмпирического метода
Total charge (Полный заряд системы) вычисляется как разность между полным количеством
Диалоговое окно полуэмпирического метода
Total charge (Полный заряд системы) вычисляется как разность между полным количеством

Слайд 12Convergence Limit (Параметр сходимости). SCF расчет заканчивается тогда, когда отличия в полной энергии двух последующих
Convergence Limit (Параметр сходимости). SCF расчет заканчивается тогда, когда отличия в полной энергии двух последующих

Слайд 13Ab initio (неэмпирические методы расчета)
Базисный набор (basic set) – это набор функций, используемых для
Ab initio (неэмпирические методы расчета)
Базисный набор (basic set) – это набор функций, используемых для

 Вклад женщин в развитие информационных технологий на рубеже XIX – XX веков
Вклад женщин в развитие информационных технологий на рубеже XIX – XX веков Подходы к понятию информации и измерению информации. Информационные объекты различных видов
Подходы к понятию информации и измерению информации. Информационные объекты различных видов Файловые системы. Жесткие диски. FAT, EXT. Особенности ФС. LVM в Linux
Файловые системы. Жесткие диски. FAT, EXT. Особенности ФС. LVM в Linux КОМПАС +. Возраст: 9 -10 класс
КОМПАС +. Возраст: 9 -10 класс Digital parenting (цифровое воспитание)
Digital parenting (цифровое воспитание) Автокодировщики. RNN, GANs
Автокодировщики. RNN, GANs Осуществление интеграции программных модулей
Осуществление интеграции программных модулей Модель Кларка – Вилсона
Модель Кларка – Вилсона IT технологии в гостницах
IT технологии в гостницах Современные инструменты для разработки и проектирования цифровой части ПС
Современные инструменты для разработки и проектирования цифровой части ПС Лаб.р.3, Тукбаева Р.В., 04.2-010
Лаб.р.3, Тукбаева Р.В., 04.2-010 Сервисы M Gmail
Сервисы M Gmail Популяризация истории среди школьников через социальные сети
Популяризация истории среди школьников через социальные сети Сохранение сметы
Сохранение сметы Алгоритм. Способы записи алгоритма
Алгоритм. Способы записи алгоритма Памятка для студентов заочной формы обучения по работе с электронными ресурсами через личный кабинет
Памятка для студентов заочной формы обучения по работе с электронными ресурсами через личный кабинет Чтобы пригласить нового пользователя
Чтобы пригласить нового пользователя Pointers in C++
Pointers in C++ Текстовый редактор SciNotes
Текстовый редактор SciNotes Программирование и алгоритмизация. Лабораторная работа №1
Программирование и алгоритмизация. Лабораторная работа №1 Распространение информации
Распространение информации Семейство SIMATIC S7
Семейство SIMATIC S7 Инициатива A
Инициатива A Портал Государственніх Услуг РФ
Портал Государственніх Услуг РФ Безопасность в сети интернет. Дистанционное воспитательное мероприятие
Безопасность в сети интернет. Дистанционное воспитательное мероприятие Основные понятия реляционной БД
Основные понятия реляционной БД Программа Full Survice
Программа Full Survice Курс Информатика
Курс Информатика