- Хромосомные заболевания человека

Содержание
- 2. Словарь Хромосомные болезни – наследственные заболевания, обусловленные изменением числа или структуры хромосом. Частота Хромосомных болезней среди
- 3. Моносомия - отсутствие в хромосомном наборе диплоидного организма одной хромосомы. Клетку или организм, у которых та
- 4. Типы наследственности 1. Аутосомно-доминантный тип наследования: а. При достаточном числе потомков признак обнаруживается в каждом поколении
- 5. Наследственные заболевания: Хромосомные болезни Болезни обмена веществ Нарушения иммунитета Болезни с преимущественным поражением эндокринной системы Болезни
- 6. Хромосомные болезни: Синдром Патау Синдром Дауна Синдром Эдвардса Синдром Шерешевского-Тернера Синдром «кошачьего крика» Синдром Клайнфельтера Синдром
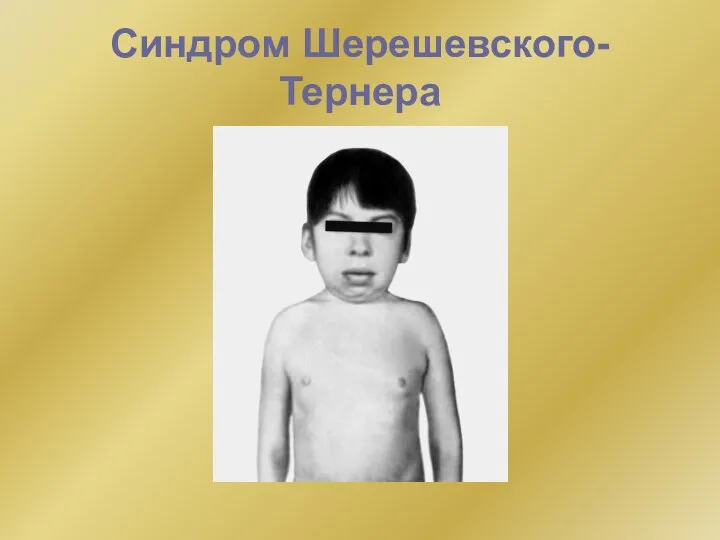
- 8. Синдром Шерешевского-Тернера Известно, что пол женщины и мужчины определяется наличием двух половых хромосом: у женщины -
- 9. Синдром Шерешевского-Тернера
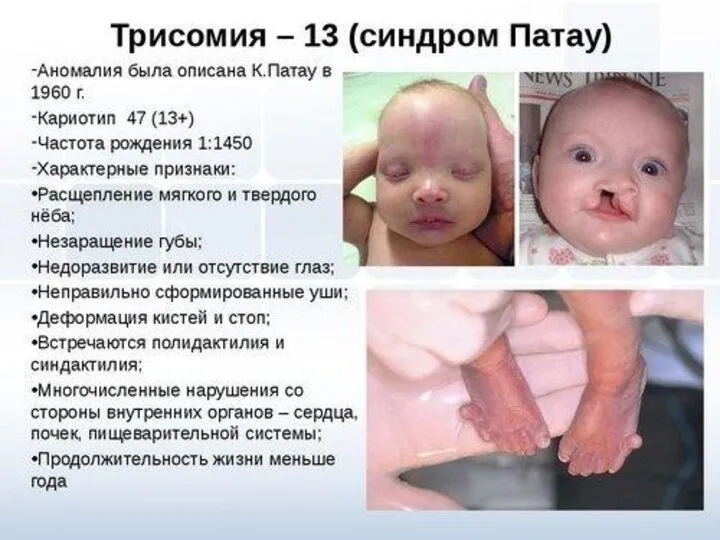
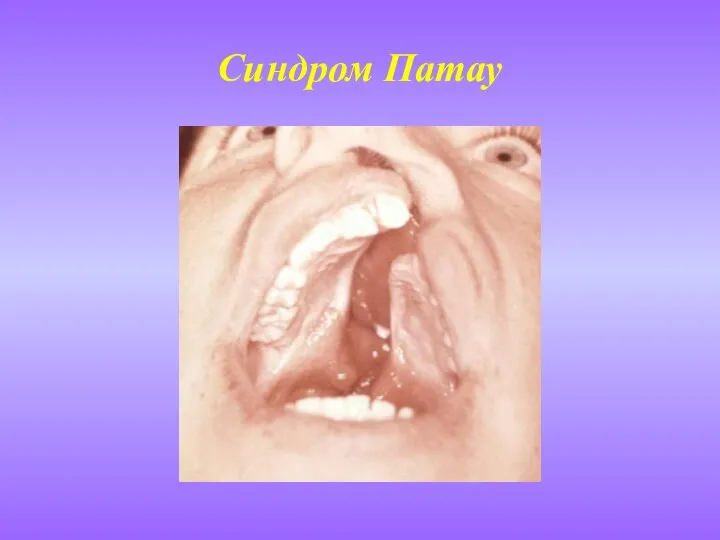
- 10. Синдром Патау (синдром 13-трисомии) Встречается примерно в одном случае на 25 тыс. живорожденных детей. Риск увеличивается
- 11. Синдром Патау
- 12. Синдром Эдвардса (синдром 18-трисомии) Встречается в одном случае на 6600 живорожденных, почти 80% пораженных — девочки.
- 13. Синдром Эдвардса
- 14. Синдром Дауна (синдром 21-трисомии) Встречается в одном случае на 7—10 тыс. живорожденных детей обоих полов во
- 15. Синдром Дауна
- 16. А теперь немного о мало известных хромосомных заболеваниях человека...
- 17. Синдром «кошачьего крика» Синдром кошачьего крика (5р-) обусловлен делецией короткого плеча 5-й хромосомы. Популяционная частота синдрома
- 18. Синдром «кошачьего крика»
- 19. Синдром Клайнфельтера Синдром Клайнфельтера включает случаи полисомии по половым хромосомам, при которых имеется не менее двух
- 20. Синдром Клайнфельтера
- 21. Синдром дубль-Y Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г. Частота синдрома
- 23. Скачать презентацию
Слайд 2Словарь
Хромосомные болезни – наследственные заболевания, обусловленные изменением числа или структуры хромосом. Частота
Словарь
Хромосомные болезни – наследственные заболевания, обусловленные изменением числа или структуры хромосом. Частота

Слайд 3Моносомия - отсутствие в хромосомном наборе диплоидного организма одной хромосомы. Клетку или
Моносомия - отсутствие в хромосомном наборе диплоидного организма одной хромосомы. Клетку или

Слайд 4Типы наследственности
1. Аутосомно-доминантный тип наследования:
а. При достаточном числе потомков признак обнаруживается
Типы наследственности
1. Аутосомно-доминантный тип наследования:
а. При достаточном числе потомков признак обнаруживается

Слайд 5Наследственные заболевания:
Хромосомные болезни
Болезни обмена веществ
Нарушения иммунитета
Болезни с преимущественным поражением эндокринной системы
Болезни крови
Нарушение
Наследственные заболевания:
Хромосомные болезни
Болезни обмена веществ
Нарушения иммунитета
Болезни с преимущественным поражением эндокринной системы
Болезни крови
Нарушение

Слайд 6Хромосомные болезни:
Синдром Патау
Синдром Дауна
Синдром Эдвардса
Синдром Шерешевского-Тернера
Синдром «кошачьего крика»
Синдром Клайнфельтера
Синдром дубль-Y
Трисомия
Хромосомные болезни:
Синдром Патау
Синдром Дауна
Синдром Эдвардса
Синдром Шерешевского-Тернера
Синдром «кошачьего крика»
Синдром Клайнфельтера
Синдром дубль-Y
Трисомия
Слайд 8Синдром Шерешевского-Тернера
Известно, что пол женщины и мужчины определяется наличием двух половых хромосом:
Синдром Шерешевского-Тернера
Известно, что пол женщины и мужчины определяется наличием двух половых хромосом:

Слайд 9Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера
Слайд 10Синдром Патау
(синдром 13-трисомии)
Встречается примерно в одном случае на 25 тыс. живорожденных
Синдром Патау
(синдром 13-трисомии)
Встречается примерно в одном случае на 25 тыс. живорожденных

Слайд 11Синдром Патау
Синдром Патау
Слайд 12Синдром Эдвардса
(синдром 18-трисомии)
Встречается в одном случае на 6600 живорожденных, почти 80% пораженных
Синдром Эдвардса
(синдром 18-трисомии)
Встречается в одном случае на 6600 живорожденных, почти 80% пораженных

Слайд 13Синдром Эдвардса
Синдром Эдвардса

Слайд 14Синдром Дауна
(синдром 21-трисомии)
Встречается в одном случае на 7—10 тыс. живорожденных детей обоих
Синдром Дауна
(синдром 21-трисомии)
Встречается в одном случае на 7—10 тыс. живорожденных детей обоих

Слайд 15Синдром Дауна
Синдром Дауна

Слайд 16А теперь немного о мало известных хромосомных заболеваниях
человека...
А теперь немного о мало известных хромосомных заболеваниях
человека...

Слайд 17Синдром «кошачьего крика»
Синдром кошачьего крика (5р-) обусловлен делецией короткого плеча 5-й хромосомы.
Синдром «кошачьего крика»
Синдром кошачьего крика (5р-) обусловлен делецией короткого плеча 5-й хромосомы.

Слайд 18Синдром «кошачьего крика»
Синдром «кошачьего крика»

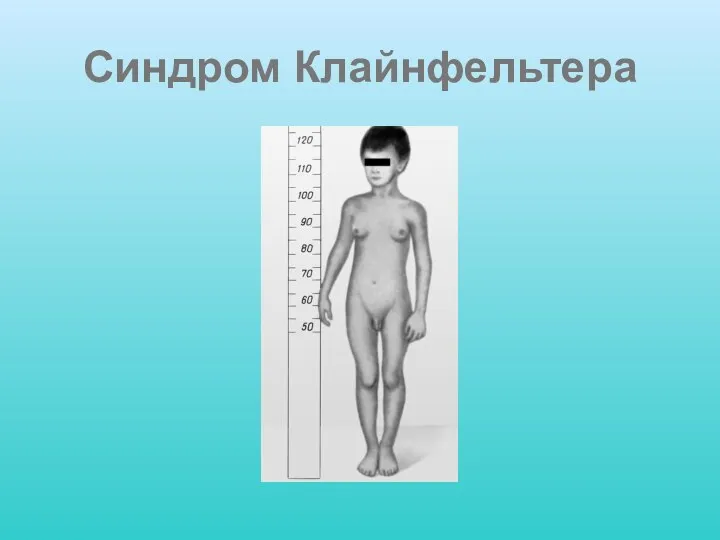
Слайд 19Синдром Клайнфельтера
Синдром Клайнфельтера включает случаи полисомии по половым хромосомам, при которых имеется
Синдром Клайнфельтера
Синдром Клайнфельтера включает случаи полисомии по половым хромосомам, при которых имеется

Слайд 20Синдром Клайнфельтера
Синдром Клайнфельтера
Слайд 21Синдром дубль-Y
Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г.
Синдром дубль-Y
Синдром XYY характеризуется кариотипом 47, XYY. Он впервые описан в 1960 г.

 Санитарно – эпидемиологический режим
Санитарно – эпидемиологический режим Синдром раздраженного кишечника
Синдром раздраженного кишечника Методы выявления туберкулеза
Методы выявления туберкулеза Аллергические заболевания и их профилактика
Аллергические заболевания и их профилактика NK-клетки (natural killer cells) и их эффекторные функции
NK-клетки (natural killer cells) и их эффекторные функции Изменение свойств пыльцы (часть 2)
Изменение свойств пыльцы (часть 2) Про туберкульоз
Про туберкульоз Майлардың алмасуынан пайда болатын аурулар
Майлардың алмасуынан пайда болатын аурулар Анатомия коленного сустава
Анатомия коленного сустава Профилактика и лечение послеоперационного гипопаратиреоза
Профилактика и лечение послеоперационного гипопаратиреоза Современные аспекты лабораторной диагностики поражений печени при хронических вирусных гепатитах В, С, D
Современные аспекты лабораторной диагностики поражений печени при хронических вирусных гепатитах В, С, D Cystic fibrosis
Cystic fibrosis Логопедические технологии формирования плавной речи
Логопедические технологии формирования плавной речи Методика для исследования мышления больных, процессов анализа и синтеза
Методика для исследования мышления больных, процессов анализа и синтеза Группы крови. Переливание крови
Группы крови. Переливание крови Хирургическая агрессология
Хирургическая агрессология Комбинированные и хронические лучевые поражения
Комбинированные и хронические лучевые поражения Шкала радиомагнитных волн
Шкала радиомагнитных волн Витамины
Витамины Профессия врача
Профессия врача Ксенобиотики и их роль в экологически обусловленных заболеваниях жителей Центрального Черноземья
Ксенобиотики и их роль в экологически обусловленных заболеваниях жителей Центрального Черноземья Гемотрансфузия
Гемотрансфузия Питание и здоровье
Питание и здоровье akro
akro Еңбек гигиенасы
Еңбек гигиенасы Проблемы сидячей работы
Проблемы сидячей работы Эффективность Гипорамина (Эребра) в лечении и профилактике вирусных заболеваний у детей
Эффективность Гипорамина (Эребра) в лечении и профилактике вирусных заболеваний у детей Гемолитическая болезнь новорожденных
Гемолитическая болезнь новорожденных