- Наследственная недостаточность Альфа-1-антитрипсина

Содержание
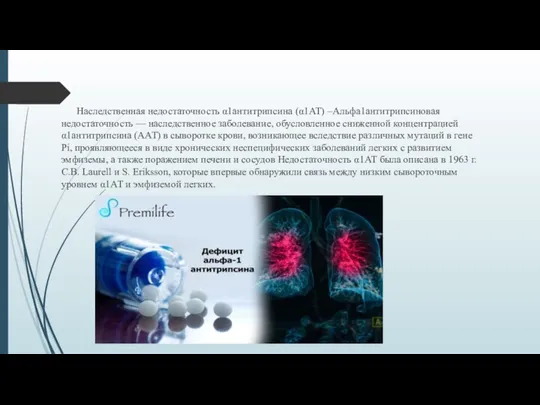
- 2. Наследственная недостаточность α1антитрипсина (α1АТ) –Альфа1антитрипсиновая недостаточность — наследственное заболевание, обусловленное сниженной концентрацией α1антитрипсина (ААТ) в сыворотке
- 3. Актуальность Принято считать, что в Европе дефицитом ААТ страдают от 1 на 1 600 до 1
- 5. Общие сведения об альфа-1-антитриписине А1АТ представляет собой низкомолекулярный гликопротеин, состоящий из 394 аминокислотных остатков и трех
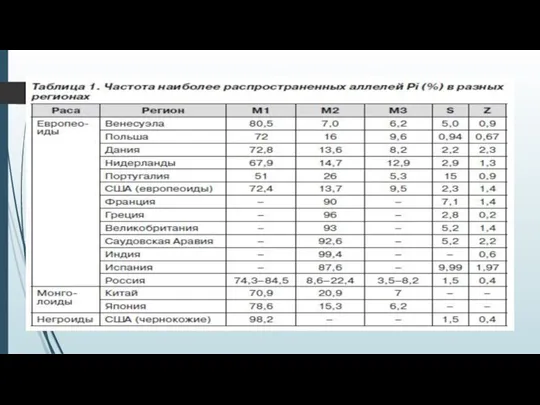
- 7. Генетический полиморфизм альфа-1-антитриписина За продукцию А1АТ отвечает ген, расположенный на хромосоме 14q32.1, называемый SERPINA1 (serpin peptidase
- 8. Варианты наследования гена альфа-1-антитрипсина ММ — оба варианта гена здоровы MS или MZ (PiMS, PiMZ) —
- 9. Есть несколько форм и степеней дефицита, которые главным образом зависят от того, сколько (1 или 2
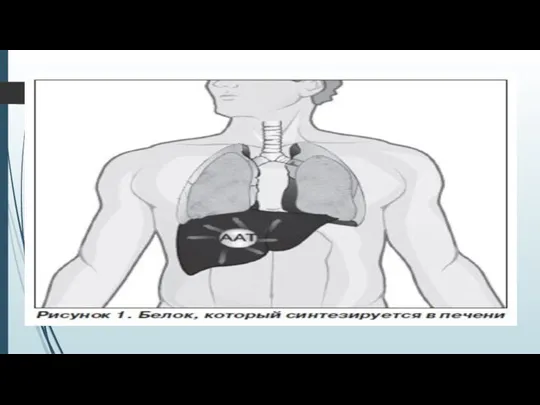
- 11. Каким образом недостаток ААТ вызывает заболевание легких? Протеин ААТ синтезируется в печени и поступает в кровь,
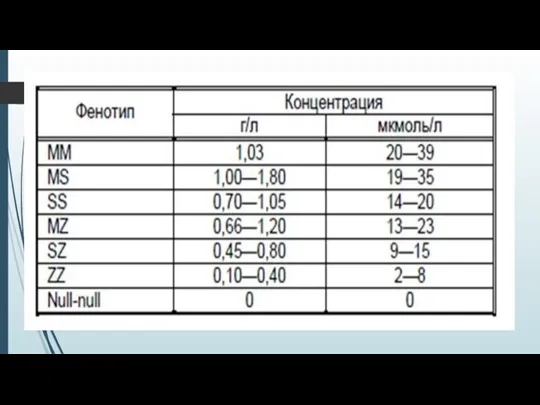
- 14. Риск развития эмфиземы значительно возрастает при снижении уровня сывороточного А1-АТ менее 0,8 г/л (11 ммоль/л) (норма
- 15. Уровень альфа-1-антитриписина в норме и физиологические колебания. Наибольшие количества А1АТ содержатся в сыворотке крови, обеспечивая 90%
- 16. Эмфизема (с греческого – «надувать, разбухать») – заболевание легких, характеризующееся патологическим расширением полостей. Легкое становится объемным
- 17. Клиника Среди основных симптомов дефицита альфа-1-антитрипсина необходимо выделить следующие: одышка, свистящее дыхание и сухие свистящие хрипы
- 18. Диагностика Заболевание диагностируется очень редко. В большинстве случаев, больные альфа-1-антитрипсином безуспешно лечатся с неверными диагнозами —
- 19. Несмотря на довольно широкую распространенность недостаточности А1АТ и несомненные преимущества ее ранней диагностики, лишь в десятке
- 20. Лечение Дефицит ААТ можно контролировать, но вылечить это заболевание невозможно. Однако ранняя диагностика имеет очень большое
- 21. Лечение дефицита альфа-1-антитрипсина Специфических методов лечения не существует. Для предупреждения прогрессирования заболевания необходимо избегать неблагоприятного воздействия
- 23. Скачать презентацию
Слайд 2 Наследственная недостаточность α1антитрипсина (α1АТ) –Альфа1антитрипсиновая недостаточность — наследственное заболевание, обусловленное сниженной
Наследственная недостаточность α1антитрипсина (α1АТ) –Альфа1антитрипсиновая недостаточность — наследственное заболевание, обусловленное сниженной
Слайд 3Актуальность
Принято считать, что в Европе дефицитом ААТ страдают от 1 на 1
Актуальность
Принято считать, что в Европе дефицитом ААТ страдают от 1 на 1

Слайд 5 Общие сведения об альфа-1-антитриписине
А1АТ представляет собой низкомолекулярный гликопротеин, состоящий из
Общие сведения об альфа-1-антитриписине
А1АТ представляет собой низкомолекулярный гликопротеин, состоящий из

Слайд 7 Генетический полиморфизм альфа-1-антитриписина
За продукцию А1АТ отвечает ген, расположенный на хромосоме
Генетический полиморфизм альфа-1-антитриписина
За продукцию А1АТ отвечает ген, расположенный на хромосоме

Слайд 8Варианты наследования гена альфа-1-антитрипсина
ММ — оба варианта гена здоровы
MS или MZ (PiMS,
Варианты наследования гена альфа-1-антитрипсина
ММ — оба варианта гена здоровы
MS или MZ (PiMS,

Слайд 9 Есть несколько форм и степеней дефицита, которые главным образом зависят от
Есть несколько форм и степеней дефицита, которые главным образом зависят от

Слайд 11Каким образом недостаток ААТ вызывает заболевание легких?
Протеин ААТ синтезируется в печени и
Каким образом недостаток ААТ вызывает заболевание легких?
Протеин ААТ синтезируется в печени и

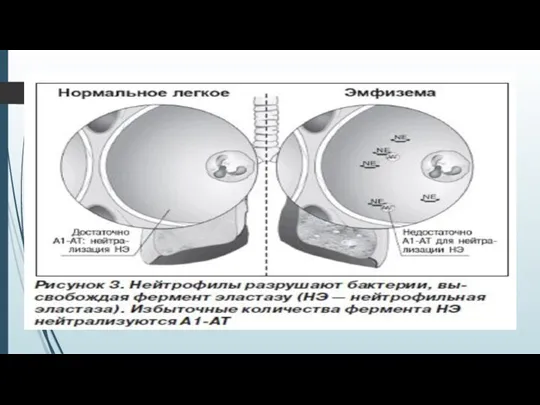

Слайд 14Риск развития эмфиземы значительно возрастает при снижении уровня сывороточного А1-АТ менее 0,8
Риск развития эмфиземы значительно возрастает при снижении уровня сывороточного А1-АТ менее 0,8

Слайд 15 Уровень альфа-1-антитриписина в норме и физиологические колебания.
Наибольшие количества А1АТ содержатся
Уровень альфа-1-антитриписина в норме и физиологические колебания.
Наибольшие количества А1АТ содержатся

Слайд 16 Эмфизема (с греческого – «надувать, разбухать») – заболевание легких, характеризующееся патологическим
Эмфизема (с греческого – «надувать, разбухать») – заболевание легких, характеризующееся патологическим

Слайд 17 Клиника
Среди основных симптомов дефицита альфа-1-антитрипсина необходимо выделить следующие: одышка, свистящее
Клиника
Среди основных симптомов дефицита альфа-1-антитрипсина необходимо выделить следующие: одышка, свистящее

Слайд 18Диагностика
Заболевание диагностируется очень редко. В большинстве случаев, больные альфа-1-антитрипсином безуспешно лечатся
Диагностика
Заболевание диагностируется очень редко. В большинстве случаев, больные альфа-1-антитрипсином безуспешно лечатся

Слайд 19Несмотря на довольно широкую распространенность недостаточности А1АТ и несомненные преимущества ее ранней
Несмотря на довольно широкую распространенность недостаточности А1АТ и несомненные преимущества ее ранней

Слайд 20Лечение
Дефицит ААТ можно контролировать, но вылечить это заболевание невозможно. Однако ранняя диагностика
Лечение
Дефицит ААТ можно контролировать, но вылечить это заболевание невозможно. Однако ранняя диагностика

Слайд 21 Лечение дефицита альфа-1-антитрипсина
Специфических методов лечения не существует. Для предупреждения прогрессирования заболевания
Лечение дефицита альфа-1-антитрипсина
Специфических методов лечения не существует. Для предупреждения прогрессирования заболевания

 топ аан нижней конечности
топ аан нижней конечности 1663560298206863
1663560298206863 Синие пороки сердца: тетрада Фалло и ТМС
Синие пороки сердца: тетрада Фалло и ТМС vvedene_v_pediatriyu
vvedene_v_pediatriyu Оптимизация параметров глубинной стимуляции мозга при нейрохирургическом лечении пациентов с дистонией
Оптимизация параметров глубинной стимуляции мозга при нейрохирургическом лечении пациентов с дистонией Противоэпидемические мероприятия в профилактике инфекционных заболеваний
Противоэпидемические мероприятия в профилактике инфекционных заболеваний Жіті инфекциялық аурулардың патологиялық морфологиясы
Жіті инфекциялық аурулардың патологиялық морфологиясы Врождёная долевая эмфизема
Врождёная долевая эмфизема Секреты здорового питания
Секреты здорового питания Стопа
Стопа Рентгенодиагностика опухолей желудочно-кишечного тракта
Рентгенодиагностика опухолей желудочно-кишечного тракта Ацетилсалициловая кислота. Механизм действия, зависимость эффекта от дозы, показания к применению, побочные эффекты
Ацетилсалициловая кислота. Механизм действия, зависимость эффекта от дозы, показания к применению, побочные эффекты Мое здоровье
Мое здоровье Кровь
Кровь Zanyatie_3_Narushenia_termoregulyatsii_Likhoradka
Zanyatie_3_Narushenia_termoregulyatsii_Likhoradka Системный остеопороз в практике врача первичного звена
Системный остеопороз в практике врача первичного звена Остеомаляция. Причины и механизмы развития остеомаляции
Остеомаляция. Причины и механизмы развития остеомаляции пороки сердца
пороки сердца Оценка качества жизни при выполнении различных видов операции экстракции катаракты с имплантацией интраокулярных линз
Оценка качества жизни при выполнении различных видов операции экстракции катаракты с имплантацией интраокулярных линз Технология использования метода переноса генов в нейроны головного мозга с помощью липосом
Технология использования метода переноса генов в нейроны головного мозга с помощью липосом Заболевания пищеварительного тракта
Заболевания пищеварительного тракта Парентеральды тамақтандыру
Парентеральды тамақтандыру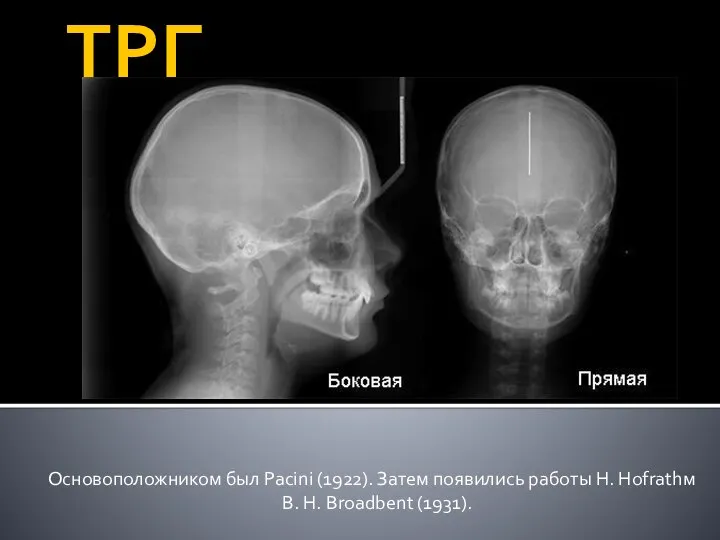 Телерентгенография
Телерентгенография Противодиабетические средства
Противодиабетические средства Теоретические основы психологии лиц с нарушениями опорно-двигательного аппарата
Теоретические основы психологии лиц с нарушениями опорно-двигательного аппарата Закупки в сфере здравоохранения в 2019 году
Закупки в сфере здравоохранения в 2019 году Мужская и женская половая система
Мужская и женская половая система Гимнастика для глаз
Гимнастика для глаз