- Наследственные болезни крови

Содержание
- 2. Наследственные болезни крови представляют собой один из важнейших разделов общей клинической гематологии. К ним, в первую
- 3. Важнейшее практическое значение в медицине имеет учение о группах крови. В связи с этим сначала рассмотрим
- 4. Группы крови АВ0 (АВН) и резус-фактор Rh
- 5. Феномен изогемагглютинации заключается в способности сыворотки крови одних людей агглютинировать или склеивать эритроциты других людей. В
- 6. Связывание эритроцитов происходит в том случае, если встречаются одноименные агглютиноген и агглютинин. В настоящее время описано
- 7. Наиболее известными из них являются группы крови системы АВ0, или ABH. В этой системе агглютиногенами являются
- 8. Антиген Н является предшественником для образования антигенов А и В. В том случае, если к антигену
- 9. Гликозилтрансфераза кодируется геном AB0, в котором идентифицированы 3 главных аллеля: A, B и 0. Аллели A
- 10. Аллель 0 — это делеция 1 нуклеотида в гене AB0, при которой фермент не синтезируется, и
- 11. Наследование групп крови по системе AB0
- 12. При этом всего могут образовываться 4 группы крови: I, или 0 при генотипе 00; II, или
- 13. Группы крови определяют иммунологические свойства агглютиногена, локализованного на поверхности эритроцитов, и взаимодействующего с ними агглютинина, растворенного
- 14. Взаимодействия между генотипами и фенотипами по системе групп крови АВ0 Группа Генотип Антигены Антитела крови эритроцитов
- 15. Первая, или нулевая группа крови встречается в различных популяциях с частотой от 30 до 40-50%. Кровь
- 16. Введениe антигенов А или В лицам с нулевой группой крови приводит к образованию антител, вызывающих агглютинацию
- 17. Вторая группа крови, или А встречается среди населения примерно с такой же частотой, что и первая,
- 18. Четвертая группа крови, или АВ является самой редкой – 3-8%. У лиц с такой группой крови
- 19. Установлено, что эритроцитарные антигены могут существовать в различных вариантах – А1, А2, А3, …, В1, В2,
- 20. Поэтому во избежание посттрансфузионных осложнений в настоящее время по жизненным показаниям разрешено переливание донорской крови только
- 21. Другая система групповых антигенов, названная системой резус-фактора (Rh), находится под более сложным генетическим контролем. Она включает
- 22. Основная роль в Rh-системе принадлежит антигену D, продукту гена RHD. При его наличии кровь является резус-положительной.
- 23. От 0,2% до 1% людей имеют особый, «слабый» вариант антигена D, обозначаемый Du. Причиной появления этого
- 24. Антигены C/c и E/e кодируются геном RHCE и образуются в результате альтернативного сплайсинга. Доля лиц с
- 25. Знание групповой принадлежности по Rh-системе имеет определяющее значение для предотвращения резус-конфликта между матерью и плодом, который
- 26. Если у резус-отрицательной женщины муж имеет резус-положительную принадлежность, то с высокой вероятностью ребенок окажется резус-положительный, и
- 27. В 15% подобных случаев после 7 недели беременности, когда в крови плода появляются зрелые эритроциты, в
- 28. Через плаценту они попадают в кровь плода и в отдельных случаях могут там накапливаться, вызывая агглютинацию
- 29. Особенно велика вероятность возникновения резус-конфликта при повторных беременностях. Во время родов или медицинском аборте кровь плода
- 30. При последующих беременностях резус-несовместимым плодом титр анти-Rh-антител в крови женщины резко возрастает. Следствием этого процесса является
- 31. При такой желтухе имеется риск формирования билирубиновой энцефалопатии, наиболее тяжелым исходом которой является детский церебральный паралич
- 32. Степень поражения ЦНС и других органов зависит от уровня непрямого билирубина, поступающего в кровь из разрушенных
- 33. Наиболее эффективным средством лечения гемолитической болезни новорожденных является обменное переливание крови в первые сутки жизни, а
- 34. Для профилактики резус-конфликта и гемолитической болезни у плода женщине с отрицательной резус-принадлежностью при любом внутриматочном вмешательстве
- 35. Введение анти-D-иммуноглобулина при повторных беременностях не показано, так как женщина уже сенсибилизирована, то есть чувствительна к
- 36. Гемоглобинопатии
- 37. Гемоглобинопатии — это гетерогенная группа наследственных заболеваний, обусловленных мутациями в глобиновых генах. В организме человека гемоглобин
- 38. 98% гемоглобина взрослого человека представлено изоформой HbA, или гемоглобином А, в состав которого входят две α-
- 39. В первой половине беременности у плода присутствуют необычные формы эмбрионального гемоглобина HbCower2 (α2; ε2) который в
- 40. В течение первого года жизни ребенка происходит замена фетального гемоглобина на гемоглобин А, и у взрослых
- 41. Гены различных цепей гемоглобина кластерированы в двух различных цитогенетических областях и являются классическим примером генных семейств,
- 42. Продукты этих генов идентичны друг другу, но характер их экспрессии различен. Ген HBA2 экспрессируется значительно интенсивнее
- 43. Остальные цепи гемоглобина (β, δ, γ и ξ) кодируются соответственно генами HBB, HBD, HBG1, HBG2 (2
- 45. Порядок локализации генов на хромосоме соответствует последовательности их экспрессии в онтогенезе, что ясно указывает на существование
- 46. При снижении экспрессии одного из генов кластера, компенсаторно повышается экспрессия других генов, и образуются гемоглобины, не
- 47. В настоящее время описано большое количество мутаций в различных генах гемоглобинов, которые приводят к развитию гемоглобинопатий
- 48. α-талассемии – это клинически полиморфная и генетически гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями в двух α-глобиновых
- 49. Гемолиз эритроцитов возникает в результате их перегрузки железом, нарушающим эритропоэз и оказывающим повреждающее действие на мембраны
- 50. У 80% больных развивается гепатоспленомегалия и у 30% - скелетные деформации. α-талассемии встречаются во многих частях
- 51. Основной тип мутаций в α-глобиновых генах (HBA1 и HBA2) — это протяженные делеции, которые могут захватывать
- 52. При этом в разных популяциях найдены специфические мажорные делеции. Среди них самыми частыми в южной Азии,
- 53. Делеция одной копии любого из двух генов, как правило, не имеет клинических проявлений. Делеция двух копий
- 54. Классическая картина хронической гемолитической анемии, сопровождающаяся образованием новых типов гемоглобина, например, гемоглобина HbH, состоящего из 4
- 55. В остальных случаях чаще всего наблюдается сочетание делеций двух копий генов с гетерозиготными точковыми мутациями в
- 56. Некоторые точковые мутации затрагивают район α-цепей, важный для осуществления их контактов с β-цепями. В этом случае
- 57. При делеции четырех копий α-глобиновых генов α-цепи полностью отсутствуют и образуется гемоглобин, состоящий из 4 γ-цепей
- 58. β-талассемии – это группа клинически полиморфных аутосомно-рецессивных заболеваний, обусловленных мутациями в β-глобиновом гене. Заболевание часто встречается
- 59. Оказалось, что в тех же регионах мира распространен малярийный плазмодий, вызывающий тяжелое протозойное заболевание – малярию.
- 60. Частота гетерозиготного носительства мутации в гене β-глобина в этих популяциях достигает 5-8%. Максимальная распространенность β-талассемии зарегистрирована
- 61. В зависимости от типа мутации и наличия β-глобиновых цепей выделяют β0- и β+-талассемию. К β0-талассемии приводят
- 62. Примером тяжелой формы β0-талассемии является анемия Кули. Болезнь проявляется в возрасте от нескольких недель до 2
- 63. При анализе крови выявляется тяжелая гипохромная анемия со снижением содержания гемоглобина и уменьшением количества эритроцитов и
- 64. Часто возникают осложнения в виде трофических язв, цирроза печени, мочекислого диатеза и гемосидероза. Причиной развития β+-талассемии
- 65. К редким вариантам β-талассемии относится персистирование фетального гемоглобина, при котором не происходит его замены на гемоглобин
- 66. Одна из миссенс-мутаций (S) в гене HBB, сопровождающаяся заменой глютамина на валин в 6 положении β-цепи,
- 67. Это приводит к дефекту мембраны эритроцитов, обуславливающему их серповидную форму. Серповидные клетки увеличивают вязкость крови и
- 68. Такие эритроциты теряют пластичность, закупоривают капилляры и гемолизируются, вызывая развитие очагов ишемии и инфаркты с выраженной
- 69. Серповидноклеточная анемия является примером наследования с неполным доминированием. Гетерозиготные носители мутации (Ss) в гене гемоглобина имеют
- 70. В обычных условиях у этих лиц анемия не выявляется, так как в их крови содержится нормальный
- 71. Наследственные анемии
- 72. Не только гемоглобинопатии, но и многие другие наследственные дефекты могут привести к развитию анемии. Аутосомно-рецессивные или
- 73. Это – глюкозо-6-фосфатдегидрогеназа (G6PD), уридин-5'-монофосфат-гидролаза (UMPH1), пируваткиназа (PKLR), фосфоглицераткиназа (PGK1), глюкозофосфатизомераза (GPI), аденилаткиназа (AK1)
- 74. Генетически гетерогенная группа Х-сцепленной и аутосомно-рецессивной анемии Фанкони характеризуется высокой геномной нестабильностью, чувствительностью к агентам, вызывающим
- 75. Анемия Фанкони, являясь одной из форм апластической анемии, клинически проявляется прогрессирующей панцитопенией и нередко сопровождается аномалиями
- 76. Одним из наиболее ранних проявлений заболевания является функциональная недостаточность костного мозга, обусловленная нарушением пролиферации и дифференцировки
- 77. В настоящее время идентифицированы 15 генов, мутации которых приводят к анемии Фанкони. Продукты этих генов являются
- 78. Центральным из этих путей является активация и транспорт к месту повреждения ДНК белков, кодируемых генами FANCD2
- 79. Мутации в гене FANCD2 являются самой частой причиной анемии Фанкони. Активация продукта гена FANCD2 происходит при
- 80. Репарация перекрестных сшивок ДНК происходит при участии многих других белков, 5 из которых кодируются генами, мутантными
- 81. Врожденная дизэритропоэтическая анемия — это гетерогенная группа заболеваний, обусловленных неэффективным эритропоэзом. При редком аутосомно-рецессивном типе I,
- 82. Причиной заболевания являются мутации в гене коданина-1 (CDAN1). Коданин-1 – это высоко консервативный белок, необходимый для
- 83. При наиболее частом II типе заболевания, характеризующимся нормоцитарными, би- или многоядерными эритроцитами с двойными цитоплазматическими мембранами,
- 84. Редкий тип III умеренной дизэритропоэтической анемии, который называется также доброкачественным эритроретикулезом, наследуется по аутосомно-доминантному типу и
- 85. Аутосомно-доминантная врожденная дизэритропоэтическая анемия IV типа, обусловленная мутациями в гене транскрипционного активатора (KLF1), по своим клиническим
- 86. Сидеробластическая анемия характеризуется присутствием в крови эритроидных сидеробластов. Ведущими клиническими проявлениями являются: гипохромная анемия, присутствие двух
- 87. Характерны системная перегрузка железом в результате хронического неэффективного эритропоэза и часто положительный гематологический ответ (ретикулоцитарный криз)
- 88. Наследственные формы заболевания включают в себя два типа — Х-сцепленную рецессивную, пиридоксин(витамин В6)-зависимую, обусловленную мутациями в
- 89. Продуктом гена ALAS2 является эритроид-специфический фермент – дельта-аминолевулинатсинтетаза, катализирующая первый шаг в биосинтезе гема
- 90. У больных с аутосомно-рецессивной формой заболевания также снижен уровень дельта-аминолевулинатсинтетазы 2 в результате недостаточности одного из
- 91. В12-дефицитная, или мегалобластная пернициозная (злокачественная) анемия обусловлена дефицитом витамина В12, который связан с его недостаточным поступлением
- 92. Редкие моногенные формы В12-дефицитной пернициозной анемии связаны с нарушением усвоения витамина В12 в организме
- 93. Поступающий с пищей витамин В12 образует комплекс с внутренним желудочным фактором, который затем взаимодействует со специфическим
- 94. Среди наследственных типов мегалобластной анемии наиболее известным является синдром Имерслунд-Гресбека, характеризующийся одновременным поражением у детей чаще
- 95. Два генетических типа заболевания – финский и норвежский – обусловлены мутациями в генах CUBN и AMN.
- 96. Более редкий генетический тип пернициозной анемии с дефицитом витамина В12 обусловлен наследственной недостаточностью внутреннего желудочного фактора,
- 97. Наследственная В12-дефицитная мегалобластная анемия, сочетающаяся с сахарным диабетом и нейросенсорной тугоухостью, является одним из проявлений синдрома
- 98. Аутосомно-рецессивная мегалобластная анемия нередко сочетается с задержкой психического развития, генерализованными судорожными приступами, развивающимися в младенческом возрасте,
- 99. Этот генетический тип мегалобластной анемии обусловлен наследственной недостаточностью ключевого фермента метаболизма фолатов – дигидрофолатредуктазы, кодируемой геном
- 100. Железодефицитная рефрактерная микроцитарная анемия без признаков нарушения метаболизма железа или кровотечений обусловлена мутациями в гене TMPRSS6.
- 101. Напомним, что мутации в гене гепсидина приводят к аутосомно-рецессивному ювенильному гемохроматозу типа 2B. Прием препаратов железа
- 102. Гетерогенная группа аутосомно-доминантных младенческих форм анемии Блекфена-Даймонда, сопровождающихся врожденной эритроидной дисплазией, обусловлена мутациями в генах различных
- 103. Характерным является хроническое течение анемии, однако у части больных, чаще в периоде пубертата, отмечается спонтанная ремиссия.
- 104. В настоящее время идентифицированы 10 генов рибосомных белков, суммарно объясняющих более 50% всех случаев анемии Блекфена-Даймонда.
- 105. Апластическая анемия — заболевание кроветворной системы, относящееся к миелодисплазиям и выражающееся в резком угнетении или прекращении
- 106. Без лечения больные с тяжелыми формами апластической анемии погибают в течение нескольких месяцев. При своевременном адекватном
- 107. Согласно современным представлениям ключевым фактором в понимании патогенеза апластической анемии является положение о главенствующей роли дефекта
- 108. Этот дефект близок по характеру или идентичен соматической мутации. Об этом свидетельствует восстановление кроветворения у больных
- 109. В 15% случаев причиной аплазии является прием лекарственных препаратов или инфекционный процесс, хотя наследственные причины подобной
- 110. В 5-10% случаев заболевание носит семейный характер или сочетается с другими соматическими аномалиями. К наследственным формам
- 111. В настоящее время идентифицированы гены, гетерозиготные мутации в которых повышают риск развития апластической анемии. Это гены
- 112. Кроме того, укорочение теломер, обусловленное присутствием гетерозиготных мутаций в генах TERT и TERC, также повышает риск
- 113. Гемофилия А и гемофилия В
- 114. В основе развития Х-сцепленных рецессивных форм гемофилии А и гемофилии В лежат наследственные дефекты двух факторов
- 115. При любой из этих форм наблюдаются нарушения свертывания крови, и самые незначительные травмы без специальной гематологической
- 116. Отметим, что у женщин – носительниц мутаций в одном из генов гемофилии в отдельных случаях также
- 117. Фактор VIII – это большой сывороточный гликопротеин, который функционирует в коагуляционном каскаде как кофактор для активации
- 118. Около 10% всех идентифицированных мутаций в гене F8С – делеции одного или нескольких смежных нуклеотидов. Остальные
- 119. Подобные инверсии полностью инактивируют ген F8С. Их возникновение связано с особенностями молекулярной структуры гена F8С
- 120. Около 14% матерей больных мальчиков являются соматическими или гонадными мозаиками, и вероятность повторного рождения больного ребенка
- 121. Фактор IX в плазме крови находится в виде гетеродимера, состоящего из двух полипептидных цепей (легкой и
- 122. Он циркулирует в виде неактивной формы до тех пор, пока не произойдет протеолитическое высвобождение активирующего его
- 123. Его роль в свертывании крови связана с активацией фактора X посредством взаимодействия с ионами кальция, фосфолипидами
- 124. Для генов F8 и F9 характерна высокая частота возникновения мутаций (4х10–6 за поколение), причем мутации значительно
- 125. При этом с возрастом вероятность возникновения новых мутаций в гене F9 у отца повышается. По разным
- 126. Точковые мутации в промоторной области гена связаны с более легкими формами заболевания, такими, например, как Лейденская
- 127. Болезнь Виллебранда
- 128. Болезнь Виллебранда — это частое наследственное заболевание крови, проявляющееся спонтанными кровотечениями. Распространенность болезни Виллебранда – 1
- 129. Основными клиническими проявлениями заболевания являются обильные носовые и десневые кровотечения, кровотечения из внутренних органов с образованием
- 130. В основе патогенеза лежат нарушения агрегации тромбоцитов, возникающие вследствие недостаточности фактора Виллебранда (ФВ) — большого мультимерного
- 131. Болезнь Виллебранда делится на 3 основных типа. При первом доброкачественном типе, который диагностируется в 60-80% случаев,
- 132. Второй тип заболевания, характеризующийся структурными нарушениями ФВ, выявляется у 10-30% больных. В этом случае у больных
- 133. Самый тяжелый третий тип заболевания обусловлен значительной количественной недостаточностью ФВ (менее 1% нормы) или его полным
- 134. Наследуется болезнь Виллебранда по аутосомно-доминантному типу с неполной пенетрантностью. Все клинические типы болезни Виллебранда обусловлены гетерозиготными
- 135. Большинство составляют миссенс-мутации, наиболее частой из которых является R1205H. На проявление мутаций могут оказывать влияние многие
- 136. Однако наибольшее модифицирующее влияние оказывает присутствие нулевой группы крови по системе АВ0. Это объясняется тем, что
- 137. Лечение при болезни Виллебранда определяется типом заболевания и выраженностью геморрагического синдрома. Так, при первом типе болезни
- 138. При тяжелых формах используют вирусинактивированные (не содержащие вирусов гепатита В и С) концентраты, включающие в себя
- 139. Наследственные тромбофилии
- 140. В настоящее время достигнут определенный прогресс в выявлении генетических факторов риска тромбофилии – нарушений гемостаза, обуславливающих
- 141. При наследственной тромбофилии значительно повышен риск развития в течение жизни венозных тромбозов и тромбоэмболии легочной артерии
- 142. К генетическим маркерам наследственной тромбофилии относят наследственные дефициты белков С, S и антитромбина, а также мутации
- 143. Протеины С и S, а также протромбин – синтезируемые в печени витамин К-зависимые сывороточные гликопротеины, являются
- 144. Протеин С активируется на поверхности эндотелия сосудов при помощи тромбин-тромбомодулинового комплекса. Действуя как сериновая протеаза, он
- 145. Белок S является кофактором активированного белка С. Фактор II свертывания крови конвертируется в тромбин фактором Xa
- 146. Активированный тромбин конвертирует фибриноген в фибрин, стимулируя тем самым агрегацию тромбоцитов, и в свою очередь активирует
- 147. Фактор V свертывания крови циркулирует в крови в неактивной форме. Он активируется тромбином и действует как
- 148. Главным ингибитором тромбина является антитромбин III, принадлежащий к суперсемейству ингибиторов сериновых протеиназ (серпинов)
- 149. Гетерозиготные мутации в генах протеина С (PROC) и S (PROS1) приводят к двум редким формам наследственной
- 150. Однако мутации в генах PROC, PROS1 и AT3D являются редкой причиной наследственных тромбофилий, так как их
- 151. Главной причиной наследственной тромбофилии является присутствие гомозиготных и гетерозиготных мутаций в генах F2 и F5. В
- 152. В результате фактор Va становится устойчивым к действию активированного белка С. Замена R506Q получила название Лейденской
- 153. Её гетерозиготное носительство повышает риск развития венозной тромбоэмболии в 5 раз, причем в этом случае тромбоз
- 154. Некоторые внешние факторы могут резко увеличивать частоту тромбозов при наличии Лейденской мутации. Это – хирургические вмешательства,
- 155. У гомозигот по Лейденской мутации, которые встречаются в популяциях значительно реже, риск тромбозов и тромбоэмболии составляет
- 156. В гене F5 описаны также и другие мутации, которые в гомозиготном или компаунд-гетерозиготном состоянии приводят к
- 157. Присутствие инактивирующих мутаций в гене F5 в компаунде с Лейденским фактором V (псевдогомозиготность по Лейденскому фактору
- 158. Спектр мутаций в гене F2 также хорошо изучен. Мажорной является замена G20210A. Носительство этой мутации увеличивает
- 159. Необходимо учитывать, что риск развития венозных тромбозов резко возрастает при сочетании наследственной предрасположенности к тромбофилии с
- 160. 1. наличие венозной тромбоэмболии в семейном анамнезе; 2. первый случай венозной тромбоэмболии в возрасте до 50
- 161. Разработаны схемы ведения и лечения беременных женщин с наследственными факторами предрасположенности к тромбофилии
- 163. Скачать презентацию
Слайд 2Наследственные болезни крови представляют собой один из важнейших разделов общей клинической гематологии.
Наследственные болезни крови представляют собой один из важнейших разделов общей клинической гематологии.

Слайд 3Важнейшее практическое значение в медицине имеет учение о группах крови.
В связи
Важнейшее практическое значение в медицине имеет учение о группах крови. В связи

Слайд 4Группы крови АВ0 (АВН) и
резус-фактор Rh
Группы крови АВ0 (АВН) и
резус-фактор Rh

Слайд 5Феномен изогемагглютинации заключается в способности сыворотки крови одних людей агглютинировать или склеивать
Феномен изогемагглютинации заключается в способности сыворотки крови одних людей агглютинировать или склеивать

Слайд 6Связывание эритроцитов происходит в том случае, если встречаются одноименные агглютиноген и агглютинин.
Связывание эритроцитов происходит в том случае, если встречаются одноименные агглютиноген и агглютинин.

Слайд 7Наиболее известными из них являются
группы крови системы АВ0, или ABH.
В
Наиболее известными из них являются группы крови системы АВ0, или ABH. В

Слайд 8Антиген Н является предшественником для образования антигенов А и В.
В том
Антиген Н является предшественником для образования антигенов А и В. В том

Слайд 9Гликозилтрансфераза кодируется геном AB0,
в котором идентифицированы
3 главных аллеля: A, B
Гликозилтрансфераза кодируется геном AB0, в котором идентифицированы 3 главных аллеля: A, B

Слайд 10Аллель 0 — это делеция 1 нуклеотида в гене AB0,
при которой
Аллель 0 — это делеция 1 нуклеотида в гене AB0, при которой

Слайд 11Наследование групп крови по системе AB0
Наследование групп крови по системе AB0
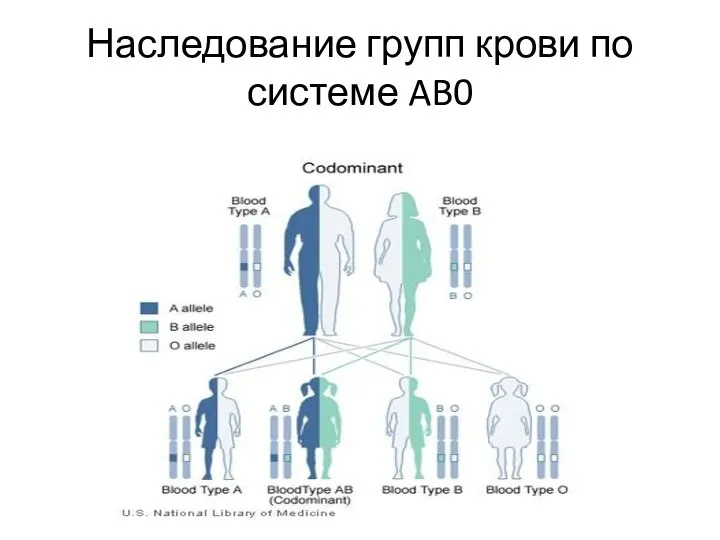
Слайд 12При этом всего могут образовываться 4 группы крови: I, или 0 при
При этом всего могут образовываться 4 группы крови: I, или 0 при

Слайд 13Группы крови определяют иммунологические свойства агглютиногена,
локализованного на поверхности эритроцитов,
и взаимодействующего
Группы крови определяют иммунологические свойства агглютиногена, локализованного на поверхности эритроцитов, и взаимодействующего

Слайд 14Взаимодействия между генотипами и фенотипами по системе групп крови АВ0
Группа
Взаимодействия между генотипами и фенотипами по системе групп крови АВ0
Группа

Слайд 15Первая, или нулевая группа крови встречается в различных популяциях с частотой от
Первая, или нулевая группа крови встречается в различных популяциях с частотой от

Слайд 16Введениe антигенов А или В лицам с нулевой группой крови приводит к
Введениe антигенов А или В лицам с нулевой группой крови приводит к

Слайд 17Вторая группа крови, или А встречается среди населения примерно с такой же
Вторая группа крови, или А встречается среди населения примерно с такой же

Слайд 18Четвертая группа крови, или АВ является самой редкой – 3-8%.
У лиц
Четвертая группа крови, или АВ является самой редкой – 3-8%. У лиц

Слайд 19Установлено, что эритроцитарные антигены могут существовать в различных вариантах –
А1, А2,
Установлено, что эритроцитарные антигены могут существовать в различных вариантах – А1, А2,

Слайд 20Поэтому во избежание посттрансфузионных осложнений в настоящее время по жизненным показаниям разрешено
Поэтому во избежание посттрансфузионных осложнений в настоящее время по жизненным показаниям разрешено

Слайд 21Другая система групповых антигенов, названная
системой резус-фактора (Rh), находится под более сложным
Другая система групповых антигенов, названная системой резус-фактора (Rh), находится под более сложным

Слайд 22Основная роль в Rh-системе принадлежит антигену D, продукту гена RHD.
При его
Основная роль в Rh-системе принадлежит антигену D, продукту гена RHD. При его

Слайд 23От 0,2% до 1% людей имеют особый, «слабый» вариант антигена D, обозначаемый
От 0,2% до 1% людей имеют особый, «слабый» вариант антигена D, обозначаемый

Слайд 24
Антигены C/c и E/e кодируются геном RHCE и образуются в результате альтернативного
Антигены C/c и E/e кодируются геном RHCE и образуются в результате альтернативного

Слайд 25Знание групповой принадлежности по Rh-системе имеет определяющее значение для предотвращения резус-конфликта между
Знание групповой принадлежности по Rh-системе имеет определяющее значение для предотвращения резус-конфликта между

Слайд 26Если у резус-отрицательной женщины муж имеет
резус-положительную принадлежность, то с высокой вероятностью
Если у резус-отрицательной женщины муж имеет резус-положительную принадлежность, то с высокой вероятностью

Слайд 27В 15% подобных случаев после
7 недели беременности, когда в крови плода
В 15% подобных случаев после 7 недели беременности, когда в крови плода

Слайд 28Через плаценту они попадают в кровь плода и в отдельных случаях могут
Через плаценту они попадают в кровь плода и в отдельных случаях могут

Слайд 29Особенно велика вероятность возникновения резус-конфликта
при повторных беременностях.
Во время родов или
Особенно велика вероятность возникновения резус-конфликта при повторных беременностях. Во время родов или

Слайд 30При последующих беременностях резус-несовместимым плодом титр анти-Rh-антител в крови женщины резко возрастает.
При последующих беременностях резус-несовместимым плодом титр анти-Rh-антител в крови женщины резко возрастает.

Слайд 31При такой желтухе имеется риск формирования билирубиновой энцефалопатии, наиболее тяжелым исходом которой
При такой желтухе имеется риск формирования билирубиновой энцефалопатии, наиболее тяжелым исходом которой

Слайд 32Степень поражения ЦНС и
других органов зависит от уровня непрямого билирубина, поступающего
Степень поражения ЦНС и других органов зависит от уровня непрямого билирубина, поступающего

Слайд 33Наиболее эффективным средством лечения гемолитической болезни новорожденных является обменное переливание крови в
Наиболее эффективным средством лечения гемолитической болезни новорожденных является обменное переливание крови в

Слайд 34Для профилактики резус-конфликта и гемолитической болезни у плода женщине с отрицательной резус-принадлежностью
Для профилактики резус-конфликта и гемолитической болезни у плода женщине с отрицательной резус-принадлежностью

Слайд 35Введение
анти-D-иммуноглобулина при повторных беременностях не показано, так как женщина уже сенсибилизирована,
Введение анти-D-иммуноглобулина при повторных беременностях не показано, так как женщина уже сенсибилизирована,

Слайд 36Гемоглобинопатии
Гемоглобинопатии

Слайд 37Гемоглобинопатии — это гетерогенная группа наследственных заболеваний, обусловленных мутациями в глобиновых генах.
Гемоглобинопатии — это гетерогенная группа наследственных заболеваний, обусловленных мутациями в глобиновых генах.

Слайд 3898% гемоглобина взрослого человека представлено изоформой HbA, или гемоглобином А, в состав
98% гемоглобина взрослого человека представлено изоформой HbA, или гемоглобином А, в состав

Слайд 39В первой половине беременности у плода присутствуют необычные формы эмбрионального гемоглобина
HbCower2 (α2;
В первой половине беременности у плода присутствуют необычные формы эмбрионального гемоглобина HbCower2 (α2;

Слайд 40В течение первого года жизни ребенка происходит замена фетального гемоглобина на гемоглобин
В течение первого года жизни ребенка происходит замена фетального гемоглобина на гемоглобин

Слайд 41Гены различных цепей гемоглобина кластерированы в двух различных цитогенетических областях и являются
Гены различных цепей гемоглобина кластерированы в двух различных цитогенетических областях и являются

Слайд 42Продукты этих генов идентичны друг другу, но характер их экспрессии различен.
Ген
Продукты этих генов идентичны друг другу, но характер их экспрессии различен. Ген

Слайд 43Остальные цепи гемоглобина
(β, δ, γ и ξ) кодируются соответственно генами
HBB,
Остальные цепи гемоглобина (β, δ, γ и ξ) кодируются соответственно генами HBB,

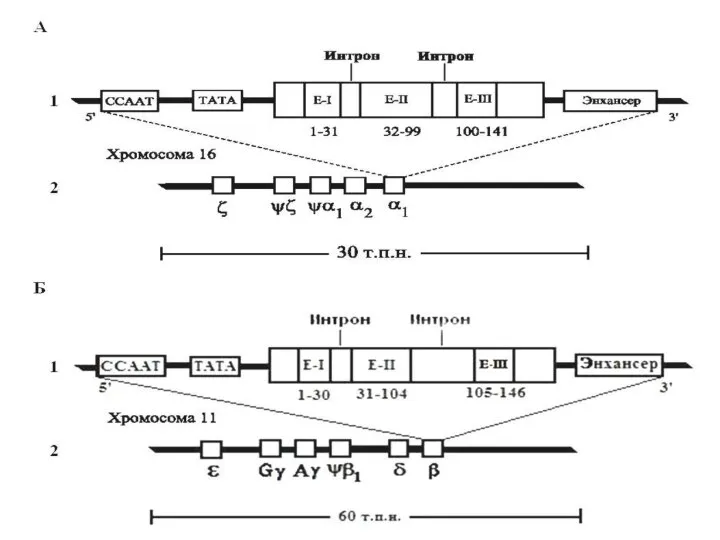
Слайд 45Порядок локализации генов на хромосоме соответствует последовательности их экспрессии в онтогенезе, что
Порядок локализации генов на хромосоме соответствует последовательности их экспрессии в онтогенезе, что

Слайд 46При снижении экспрессии одного из генов кластера, компенсаторно повышается экспрессия других генов,
При снижении экспрессии одного из генов кластера, компенсаторно повышается экспрессия других генов,

Слайд 47В настоящее время описано большое количество мутаций в различных генах гемоглобинов, которые
В настоящее время описано большое количество мутаций в различных генах гемоглобинов, которые

Слайд 48α-талассемии – это клинически полиморфная и генетически гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных
α-талассемии – это клинически полиморфная и генетически гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных

Слайд 49Гемолиз эритроцитов возникает в результате их перегрузки железом, нарушающим эритропоэз и оказывающим
Гемолиз эритроцитов возникает в результате их перегрузки железом, нарушающим эритропоэз и оказывающим

Слайд 50У 80% больных развивается гепатоспленомегалия и у 30% - скелетные деформации.
α-талассемии
У 80% больных развивается гепатоспленомегалия и у 30% - скелетные деформации. α-талассемии

Слайд 51Основной тип мутаций в α-глобиновых генах (HBA1 и HBA2) — это протяженные
Основной тип мутаций в α-глобиновых генах (HBA1 и HBA2) — это протяженные

Слайд 52При этом в разных популяциях найдены специфические мажорные делеции.
Среди них самыми
При этом в разных популяциях найдены специфические мажорные делеции. Среди них самыми

Слайд 53Делеция одной копии любого из двух генов, как правило, не имеет клинических
Делеция одной копии любого из двух генов, как правило, не имеет клинических

Слайд 54Классическая картина хронической гемолитической анемии, сопровождающаяся образованием новых типов гемоглобина, например, гемоглобина
Классическая картина хронической гемолитической анемии, сопровождающаяся образованием новых типов гемоглобина, например, гемоглобина

Слайд 55В остальных случаях чаще всего наблюдается сочетание делеций двух копий генов с
В остальных случаях чаще всего наблюдается сочетание делеций двух копий генов с

Слайд 56Некоторые точковые мутации затрагивают район α-цепей, важный для осуществления их контактов с
Некоторые точковые мутации затрагивают район α-цепей, важный для осуществления их контактов с

Слайд 57При делеции четырех копий α-глобиновых генов α-цепи полностью отсутствуют и образуется гемоглобин,
При делеции четырех копий α-глобиновых генов α-цепи полностью отсутствуют и образуется гемоглобин,

Слайд 58β-талассемии – это группа клинически полиморфных аутосомно-рецессивных заболеваний, обусловленных мутациями в β-глобиновом
β-талассемии – это группа клинически полиморфных аутосомно-рецессивных заболеваний, обусловленных мутациями в β-глобиновом

Слайд 59Оказалось, что в тех же регионах мира распространен малярийный плазмодий, вызывающий тяжелое
Оказалось, что в тех же регионах мира распространен малярийный плазмодий, вызывающий тяжелое

Слайд 60Частота гетерозиготного носительства мутации в гене
β-глобина в этих популяциях достигает 5-8%.
Частота гетерозиготного носительства мутации в гене β-глобина в этих популяциях достигает 5-8%.

Слайд 61В зависимости от типа мутации и наличия β-глобиновых цепей выделяют β0- и
В зависимости от типа мутации и наличия β-глобиновых цепей выделяют β0- и

Слайд 62Примером тяжелой формы
β0-талассемии является
анемия Кули.
Болезнь проявляется в возрасте от
Примером тяжелой формы β0-талассемии является анемия Кули. Болезнь проявляется в возрасте от

Слайд 63При анализе крови выявляется тяжелая гипохромная анемия со снижением содержания гемоглобина и
При анализе крови выявляется тяжелая гипохромная анемия со снижением содержания гемоглобина и

Слайд 64Часто возникают осложнения в виде трофических язв, цирроза печени, мочекислого диатеза и
Часто возникают осложнения в виде трофических язв, цирроза печени, мочекислого диатеза и

Слайд 65К редким вариантам β-талассемии относится персистирование фетального гемоглобина,
при котором не происходит
К редким вариантам β-талассемии относится персистирование фетального гемоглобина, при котором не происходит

Слайд 66Одна из миссенс-мутаций (S) в гене HBB, сопровождающаяся заменой глютамина на валин
Одна из миссенс-мутаций (S) в гене HBB, сопровождающаяся заменой глютамина на валин

Слайд 67Это приводит к дефекту мембраны эритроцитов, обуславливающему их серповидную форму. Серповидные клетки
Это приводит к дефекту мембраны эритроцитов, обуславливающему их серповидную форму. Серповидные клетки

Слайд 68Такие эритроциты теряют пластичность, закупоривают капилляры и гемолизируются, вызывая развитие очагов ишемии
Такие эритроциты теряют пластичность, закупоривают капилляры и гемолизируются, вызывая развитие очагов ишемии

Слайд 69Серповидноклеточная анемия является примером наследования с неполным доминированием. Гетерозиготные носители мутации (Ss)
Серповидноклеточная анемия является примером наследования с неполным доминированием. Гетерозиготные носители мутации (Ss)

Слайд 70В обычных условиях у этих лиц анемия не выявляется, так как в
В обычных условиях у этих лиц анемия не выявляется, так как в

Слайд 71Наследственные анемии
Наследственные анемии

Слайд 72Не только гемоглобинопатии, но и многие другие наследственные дефекты могут привести к
Не только гемоглобинопатии, но и многие другие наследственные дефекты могут привести к

Слайд 73Это – глюкозо-6-фосфатдегидрогеназа (G6PD), уридин-5'-монофосфат-гидролаза (UMPH1), пируваткиназа (PKLR), фосфоглицераткиназа (PGK1), глюкозофосфатизомераза (GPI),
Это – глюкозо-6-фосфатдегидрогеназа (G6PD), уридин-5'-монофосфат-гидролаза (UMPH1), пируваткиназа (PKLR), фосфоглицераткиназа (PGK1), глюкозофосфатизомераза (GPI),

Слайд 74Генетически гетерогенная группа Х-сцепленной и аутосомно-рецессивной анемии Фанкони характеризуется высокой геномной нестабильностью,
Генетически гетерогенная группа Х-сцепленной и аутосомно-рецессивной анемии Фанкони характеризуется высокой геномной нестабильностью,

Слайд 75Анемия Фанкони, являясь одной из форм апластической анемии, клинически проявляется прогрессирующей панцитопенией
Анемия Фанкони, являясь одной из форм апластической анемии, клинически проявляется прогрессирующей панцитопенией

Слайд 76Одним из наиболее ранних проявлений заболевания является функциональная недостаточность костного мозга, обусловленная
Одним из наиболее ранних проявлений заболевания является функциональная недостаточность костного мозга, обусловленная

Слайд 77В настоящее время идентифицированы 15 генов, мутации которых приводят к анемии Фанкони.
В настоящее время идентифицированы 15 генов, мутации которых приводят к анемии Фанкони.

Слайд 78Центральным из этих путей является
активация и транспорт
к месту повреждения ДНК
Центральным из этих путей является активация и транспорт к месту повреждения ДНК

Слайд 79Мутации в гене FANCD2 являются самой частой причиной анемии Фанкони.
Активация продукта
Мутации в гене FANCD2 являются самой частой причиной анемии Фанкони. Активация продукта

Слайд 80Репарация перекрестных сшивок ДНК
происходит при участии многих других белков, 5 из
Репарация перекрестных сшивок ДНК происходит при участии многих других белков, 5 из

Слайд 81Врожденная дизэритропоэтическая анемия — это гетерогенная группа заболеваний, обусловленных неэффективным эритропоэзом.
При
Врожденная дизэритропоэтическая анемия — это гетерогенная группа заболеваний, обусловленных неэффективным эритропоэзом. При

Слайд 82Причиной заболевания являются мутации в гене коданина-1 (CDAN1).
Коданин-1 – это высоко
Причиной заболевания являются мутации в гене коданина-1 (CDAN1). Коданин-1 – это высоко

Слайд 83При наиболее частом II типе заболевания, характеризующимся нормоцитарными, би- или многоядерными эритроцитами
При наиболее частом II типе заболевания, характеризующимся нормоцитарными, би- или многоядерными эритроцитами

Слайд 84Редкий тип III умеренной дизэритропоэтической анемии, который называется также доброкачественным эритроретикулезом, наследуется
Редкий тип III умеренной дизэритропоэтической анемии, который называется также доброкачественным эритроретикулезом, наследуется

Слайд 85Аутосомно-доминантная врожденная дизэритропоэтическая анемия
IV типа, обусловленная мутациями в гене транскрипционного активатора
Аутосомно-доминантная врожденная дизэритропоэтическая анемия IV типа, обусловленная мутациями в гене транскрипционного активатора

Слайд 86Сидеробластическая анемия характеризуется присутствием в крови эритроидных сидеробластов. Ведущими клиническими проявлениями являются:
Сидеробластическая анемия характеризуется присутствием в крови эритроидных сидеробластов. Ведущими клиническими проявлениями являются:

Слайд 87Характерны системная перегрузка железом в результате хронического неэффективного эритропоэза и часто положительный
Характерны системная перегрузка железом в результате хронического неэффективного эритропоэза и часто положительный

Слайд 88Наследственные формы заболевания включают в себя два типа — Х-сцепленную рецессивную, пиридоксин(витамин
Наследственные формы заболевания включают в себя два типа — Х-сцепленную рецессивную, пиридоксин(витамин

Слайд 89Продуктом гена ALAS2 является эритроид-специфический фермент – дельта-аминолевулинатсинтетаза, катализирующая первый шаг в
Продуктом гена ALAS2 является эритроид-специфический фермент – дельта-аминолевулинатсинтетаза, катализирующая первый шаг в

Слайд 90У больных с аутосомно-рецессивной формой заболевания также снижен уровень дельта-аминолевулинатсинтетазы 2
в
У больных с аутосомно-рецессивной формой заболевания также снижен уровень дельта-аминолевулинатсинтетазы 2 в

Слайд 91В12-дефицитная, или мегалобластная пернициозная (злокачественная) анемия обусловлена дефицитом витамина В12, который связан
В12-дефицитная, или мегалобластная пернициозная (злокачественная) анемия обусловлена дефицитом витамина В12, который связан

Слайд 92Редкие моногенные формы В12-дефицитной пернициозной анемии связаны с нарушением усвоения витамина В12
Редкие моногенные формы В12-дефицитной пернициозной анемии связаны с нарушением усвоения витамина В12

Слайд 93Поступающий с пищей витамин В12 образует комплекс с внутренним желудочным фактором, который
Поступающий с пищей витамин В12 образует комплекс с внутренним желудочным фактором, который

Слайд 94Среди наследственных типов мегалобластной анемии наиболее известным является
синдром Имерслунд-Гресбека, характеризующийся одновременным
Среди наследственных типов мегалобластной анемии наиболее известным является синдром Имерслунд-Гресбека, характеризующийся одновременным

Слайд 95Два генетических типа заболевания – финский и норвежский – обусловлены мутациями в
Два генетических типа заболевания – финский и норвежский – обусловлены мутациями в

Слайд 96Более редкий генетический тип пернициозной анемии с дефицитом витамина В12 обусловлен наследственной
Более редкий генетический тип пернициозной анемии с дефицитом витамина В12 обусловлен наследственной

Слайд 97Наследственная В12-дефицитная мегалобластная анемия, сочетающаяся с сахарным диабетом и нейросенсорной тугоухостью, является
Наследственная В12-дефицитная мегалобластная анемия, сочетающаяся с сахарным диабетом и нейросенсорной тугоухостью, является

Слайд 98Аутосомно-рецессивная мегалобластная анемия нередко сочетается с задержкой психического развития, генерализованными судорожными приступами,
Аутосомно-рецессивная мегалобластная анемия нередко сочетается с задержкой психического развития, генерализованными судорожными приступами,

Слайд 99Этот генетический тип мегалобластной анемии обусловлен наследственной недостаточностью ключевого фермента
метаболизма фолатов
Этот генетический тип мегалобластной анемии обусловлен наследственной недостаточностью ключевого фермента метаболизма фолатов

Слайд 100Железодефицитная рефрактерная микроцитарная анемия без признаков нарушения метаболизма железа или кровотечений обусловлена
Железодефицитная рефрактерная микроцитарная анемия без признаков нарушения метаболизма железа или кровотечений обусловлена

Слайд 101Напомним, что мутации в гене гепсидина приводят к аутосомно-рецессивному ювенильному гемохроматозу типа
Напомним, что мутации в гене гепсидина приводят к аутосомно-рецессивному ювенильному гемохроматозу типа

Слайд 102Гетерогенная группа аутосомно-доминантных младенческих форм анемии Блекфена-Даймонда, сопровождающихся врожденной эритроидной дисплазией, обусловлена
Гетерогенная группа аутосомно-доминантных младенческих форм анемии Блекфена-Даймонда, сопровождающихся врожденной эритроидной дисплазией, обусловлена

Слайд 103Характерным является хроническое течение анемии, однако у части больных, чаще в периоде
Характерным является хроническое течение анемии, однако у части больных, чаще в периоде

Слайд 104В настоящее время идентифицированы 10 генов рибосомных белков, суммарно объясняющих более 50%
В настоящее время идентифицированы 10 генов рибосомных белков, суммарно объясняющих более 50%

Слайд 105Апластическая анемия — заболевание кроветворной системы, относящееся к миелодисплазиям и выражающееся в
Апластическая анемия — заболевание кроветворной системы, относящееся к миелодисплазиям и выражающееся в

Слайд 106Без лечения больные с тяжелыми формами апластической анемии погибают в течение нескольких
Без лечения больные с тяжелыми формами апластической анемии погибают в течение нескольких

Слайд 107Согласно современным представлениям ключевым фактором в понимании патогенеза апластической анемии является положение
Согласно современным представлениям ключевым фактором в понимании патогенеза апластической анемии является положение

Слайд 108Этот дефект близок по характеру или идентичен соматической мутации. Об этом свидетельствует
Этот дефект близок по характеру или идентичен соматической мутации. Об этом свидетельствует

Слайд 109В 15% случаев причиной аплазии является прием лекарственных препаратов или инфекционный процесс,
В 15% случаев причиной аплазии является прием лекарственных препаратов или инфекционный процесс,

Слайд 110В 5-10% случаев заболевание носит семейный характер или сочетается с другими соматическими
В 5-10% случаев заболевание носит семейный характер или сочетается с другими соматическими

Слайд 111В настоящее время идентифицированы гены, гетерозиготные мутации в которых повышают риск развития
В настоящее время идентифицированы гены, гетерозиготные мутации в которых повышают риск развития

Слайд 112Кроме того, укорочение теломер, обусловленное присутствием гетерозиготных мутаций в генах TERT и
Кроме того, укорочение теломер, обусловленное присутствием гетерозиготных мутаций в генах TERT и

Слайд 113Гемофилия А и гемофилия В
Гемофилия А и гемофилия В

Слайд 114В основе развития Х-сцепленных рецессивных форм гемофилии А и гемофилии В лежат
В основе развития Х-сцепленных рецессивных форм гемофилии А и гемофилии В лежат

Слайд 115При любой из этих форм наблюдаются нарушения свертывания крови, и самые незначительные
При любой из этих форм наблюдаются нарушения свертывания крови, и самые незначительные

Слайд 116Отметим, что у женщин – носительниц мутаций в одном из генов гемофилии
Отметим, что у женщин – носительниц мутаций в одном из генов гемофилии

Слайд 117Фактор VIII – это большой сывороточный гликопротеин, который функционирует в коагуляционном каскаде
Фактор VIII – это большой сывороточный гликопротеин, который функционирует в коагуляционном каскаде

Слайд 118Около 10% всех идентифицированных мутаций в гене F8С – делеции одного или
Около 10% всех идентифицированных мутаций в гене F8С – делеции одного или

Слайд 119Подобные инверсии полностью инактивируют ген F8С.
Их возникновение связано с особенностями молекулярной
Подобные инверсии полностью инактивируют ген F8С. Их возникновение связано с особенностями молекулярной

Слайд 120Около 14% матерей больных мальчиков являются соматическими или гонадными мозаиками, и вероятность
Около 14% матерей больных мальчиков являются соматическими или гонадными мозаиками, и вероятность

Слайд 121Фактор IX в плазме крови находится в виде гетеродимера, состоящего из двух
Фактор IX в плазме крови находится в виде гетеродимера, состоящего из двух

Слайд 122Он циркулирует в виде неактивной формы до тех пор, пока не произойдет
Он циркулирует в виде неактивной формы до тех пор, пока не произойдет

Слайд 123Его роль в свертывании крови связана с активацией фактора X посредством взаимодействия
Его роль в свертывании крови связана с активацией фактора X посредством взаимодействия

Слайд 124Для генов F8 и F9 характерна высокая частота возникновения мутаций
(4х10–6 за
Для генов F8 и F9 характерна высокая частота возникновения мутаций (4х10–6 за

Слайд 125При этом с возрастом вероятность возникновения новых мутаций в гене F9 у
При этом с возрастом вероятность возникновения новых мутаций в гене F9 у

Слайд 126Точковые мутации в промоторной области гена связаны с более легкими формами заболевания,
Точковые мутации в промоторной области гена связаны с более легкими формами заболевания,

Слайд 127Болезнь Виллебранда
Болезнь Виллебранда

Слайд 128Болезнь Виллебранда —
это частое наследственное заболевание крови, проявляющееся спонтанными кровотечениями. Распространенность
Болезнь Виллебранда — это частое наследственное заболевание крови, проявляющееся спонтанными кровотечениями. Распространенность

Слайд 129Основными клиническими проявлениями заболевания являются обильные носовые и десневые кровотечения, кровотечения из
Основными клиническими проявлениями заболевания являются обильные носовые и десневые кровотечения, кровотечения из

Слайд 130В основе патогенеза лежат нарушения агрегации тромбоцитов, возникающие вследствие недостаточности
фактора Виллебранда
В основе патогенеза лежат нарушения агрегации тромбоцитов, возникающие вследствие недостаточности фактора Виллебранда

Слайд 131Болезнь Виллебранда делится на 3 основных типа.
При первом доброкачественном типе, который
Болезнь Виллебранда делится на 3 основных типа. При первом доброкачественном типе, который

Слайд 132Второй тип заболевания, характеризующийся структурными нарушениями ФВ, выявляется у
10-30% больных.
В
Второй тип заболевания, характеризующийся структурными нарушениями ФВ, выявляется у 10-30% больных. В

Слайд 133Самый тяжелый третий тип заболевания обусловлен значительной количественной недостаточностью ФВ (менее 1%
Самый тяжелый третий тип заболевания обусловлен значительной количественной недостаточностью ФВ (менее 1%

Слайд 134Наследуется болезнь Виллебранда по аутосомно-доминантному типу с неполной пенетрантностью.
Все клинические типы
Наследуется болезнь Виллебранда по аутосомно-доминантному типу с неполной пенетрантностью. Все клинические типы

Слайд 135Большинство составляют миссенс-мутации, наиболее частой из которых является R1205H.
На проявление мутаций
Большинство составляют миссенс-мутации, наиболее частой из которых является R1205H. На проявление мутаций

Слайд 136Однако наибольшее модифицирующее влияние оказывает присутствие нулевой группы крови по системе АВ0.
Однако наибольшее модифицирующее влияние оказывает присутствие нулевой группы крови по системе АВ0.

Слайд 137Лечение при болезни Виллебранда определяется типом заболевания и выраженностью геморрагического синдрома.
Так,
Лечение при болезни Виллебранда определяется типом заболевания и выраженностью геморрагического синдрома. Так,

Слайд 138При тяжелых формах используют вирусинактивированные
(не содержащие вирусов
гепатита В и С)
При тяжелых формах используют вирусинактивированные (не содержащие вирусов гепатита В и С)

Слайд 139Наследственные тромбофилии
Наследственные тромбофилии

Слайд 140В настоящее время достигнут определенный прогресс в выявлении генетических факторов риска тромбофилии
В настоящее время достигнут определенный прогресс в выявлении генетических факторов риска тромбофилии

Слайд 141При наследственной тромбофилии значительно повышен риск развития в течение жизни венозных тромбозов
При наследственной тромбофилии значительно повышен риск развития в течение жизни венозных тромбозов

Слайд 142К генетическим маркерам наследственной тромбофилии относят наследственные дефициты
белков С, S и
К генетическим маркерам наследственной тромбофилии относят наследственные дефициты белков С, S и

Слайд 143Протеины С и S, а также протромбин – синтезируемые в печени витамин
Протеины С и S, а также протромбин – синтезируемые в печени витамин

Слайд 144Протеин С активируется на поверхности эндотелия сосудов при помощи
тромбин-тромбомодулинового комплекса.
Действуя
Протеин С активируется на поверхности эндотелия сосудов при помощи тромбин-тромбомодулинового комплекса. Действуя

Слайд 145Белок S является кофактором активированного белка С.
Фактор II свертывания крови конвертируется
Белок S является кофактором активированного белка С. Фактор II свертывания крови конвертируется

Слайд 146Активированный тромбин конвертирует фибриноген в фибрин, стимулируя тем самым агрегацию тромбоцитов, и
Активированный тромбин конвертирует фибриноген в фибрин, стимулируя тем самым агрегацию тромбоцитов, и

Слайд 147Фактор V свертывания крови циркулирует в крови в неактивной форме.
Он активируется
Фактор V свертывания крови циркулирует в крови в неактивной форме. Он активируется

Слайд 148Главным ингибитором тромбина является антитромбин III, принадлежащий к суперсемейству ингибиторов сериновых протеиназ
Главным ингибитором тромбина является антитромбин III, принадлежащий к суперсемейству ингибиторов сериновых протеиназ

Слайд 149Гетерозиготные мутации в генах протеина С (PROC) и S (PROS1) приводят к
Гетерозиготные мутации в генах протеина С (PROC) и S (PROS1) приводят к

Слайд 150Однако мутации в генах
PROC, PROS1 и AT3D
являются редкой причиной наследственных
Однако мутации в генах PROC, PROS1 и AT3D являются редкой причиной наследственных

Слайд 151Главной причиной наследственной тромбофилии является присутствие гомозиготных и гетерозиготных мутаций в генах
Главной причиной наследственной тромбофилии является присутствие гомозиготных и гетерозиготных мутаций в генах

Слайд 152В результате фактор Va становится устойчивым к действию активированного белка С.
Замена
В результате фактор Va становится устойчивым к действию активированного белка С. Замена

Слайд 153Её гетерозиготное носительство повышает риск развития венозной тромбоэмболии в 5 раз, причем
Её гетерозиготное носительство повышает риск развития венозной тромбоэмболии в 5 раз, причем

Слайд 154Некоторые внешние факторы могут резко увеличивать частоту тромбозов при наличии Лейденской мутации.
Некоторые внешние факторы могут резко увеличивать частоту тромбозов при наличии Лейденской мутации.

Слайд 155У гомозигот по Лейденской мутации, которые встречаются в популяциях значительно реже, риск
У гомозигот по Лейденской мутации, которые встречаются в популяциях значительно реже, риск

Слайд 156В гене F5 описаны также и другие мутации, которые в гомозиготном или
В гене F5 описаны также и другие мутации, которые в гомозиготном или

Слайд 157Присутствие инактивирующих мутаций в гене F5 в компаунде с Лейденским фактором V
Присутствие инактивирующих мутаций в гене F5 в компаунде с Лейденским фактором V

Слайд 158Спектр мутаций в гене F2 также хорошо изучен.
Мажорной является замена G20210A.
Спектр мутаций в гене F2 также хорошо изучен. Мажорной является замена G20210A.

Слайд 159Необходимо учитывать, что риск развития венозных тромбозов резко возрастает при сочетании наследственной
Необходимо учитывать, что риск развития венозных тромбозов резко возрастает при сочетании наследственной

Слайд 1601. наличие венозной тромбоэмболии в семейном анамнезе;
2. первый случай венозной тромбоэмболии
1. наличие венозной тромбоэмболии в семейном анамнезе; 2. первый случай венозной тромбоэмболии

Слайд 161 Разработаны схемы ведения и лечения беременных женщин с наследственными факторами предрасположенности к
Разработаны схемы ведения и лечения беременных женщин с наследственными факторами предрасположенности к

 Паратонзиллярный абсцесс. Лечение, симптомы, осложнения
Паратонзиллярный абсцесс. Лечение, симптомы, осложнения Вертикальное движение нижней челюсти. Траектория смещения подбородочной точки и головок нижней челюсти
Вертикальное движение нижней челюсти. Траектория смещения подбородочной точки и головок нижней челюсти Стоматологияда жедел жағдайларының пайда болу механизмдері
Стоматологияда жедел жағдайларының пайда болу механизмдері Здоровое питание
Здоровое питание Экзогенный тип гипоксии. Гипоксическая и гипероксическая. Патогенез
Экзогенный тип гипоксии. Гипоксическая и гипероксическая. Патогенез История медицины как наука и предмет преподавания
История медицины как наука и предмет преподавания Функціональний протез руки для людей з ектродактилією
Функціональний протез руки для людей з ектродактилією Синдром диссеминированного внутрисосудистого свертывания крови (ДВС)
Синдром диссеминированного внутрисосудистого свертывания крови (ДВС) Fiche Métier Infirmière
Fiche Métier Infirmière Операция Льюиса при опухоли пищевода
Операция Льюиса при опухоли пищевода Дезинфекция. Санитарный режим медицинских организаций
Дезинфекция. Санитарный режим медицинских организаций Учение об инфекции. Биологический метод исследования
Учение об инфекции. Биологический метод исследования Наркомания стала реальностью
Наркомания стала реальностью Особенности функционального развития детей (младшего и старшего школьного возраста)
Особенности функционального развития детей (младшего и старшего школьного возраста) Синдром поражения таламуса и внутренней капсулы
Синдром поражения таламуса и внутренней капсулы Антиаллергические лекарственные препараты
Антиаллергические лекарственные препараты Отчет по выполнению индивидуального учебного плана
Отчет по выполнению индивидуального учебного плана Фазы биотрансформации лекарственных средств
Фазы биотрансформации лекарственных средств Обязательное медицинское освидетельствование иностранных студентов
Обязательное медицинское освидетельствование иностранных студентов Гемостаздық жүйенің корсеткіштері
Гемостаздық жүйенің корсеткіштері Топографическая анатомия области головы
Топографическая анатомия области головы Modern COVID-19. Vaccines. Kimberly Meg Pereira (ОрГМУ) 309и
Modern COVID-19. Vaccines. Kimberly Meg Pereira (ОрГМУ) 309и Стилізація природних форм
Стилізація природних форм ПМПк как ресурс развития профессиональной компетентности специалистов сопровождения
ПМПк как ресурс развития профессиональной компетентности специалистов сопровождения Иммунологиялық әдіс
Иммунологиялық әдіс Лептоспироз 2020 (1)
Лептоспироз 2020 (1) Сдача анализа кала
Сдача анализа кала Синдром красного глаза. Конъюктивиты. Лекция 4
Синдром красного глаза. Конъюктивиты. Лекция 4