- Familial Hypercholesterolemia

Содержание
- 2. Introduction Familial hypercholesterolemia (FH) have raised cholesterol levels in blood with a significant risk of developing
- 3. CLINICAL MANIFESTATIONS High cholesterol level in blood. Heterozygotes may have premature cardiovascular disease at the age
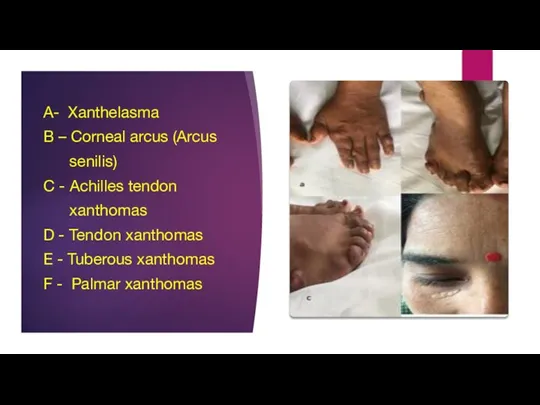
- 4. A- Xanthelasma B – Corneal arcus (Arcus senilis) C - Achilles tendon xanthomas D - Tendon
- 5. PLASMA CHOLESTEROL LEVEL IN NORMAL AND FH INDIVIDUALS NORMAL – 150 – 200 mg/dl FH HETEROZYTOGE
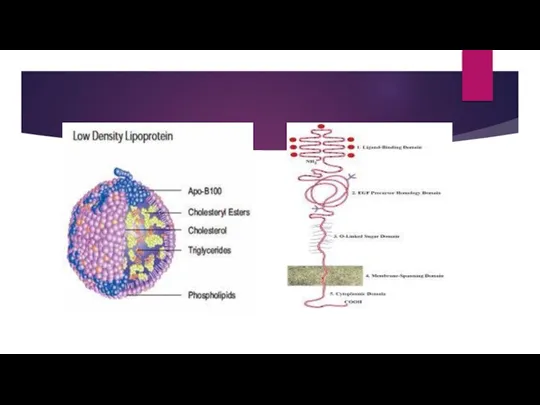
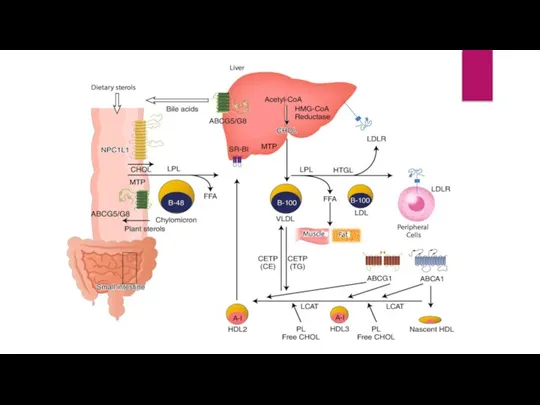
- 6. Function of LDLR gene The LDLR gene provides instructions for making a protein called low density
- 7. Mutation in LDLR gene Mutations in the LDLR gene cause FH More than 1,000 mutations have
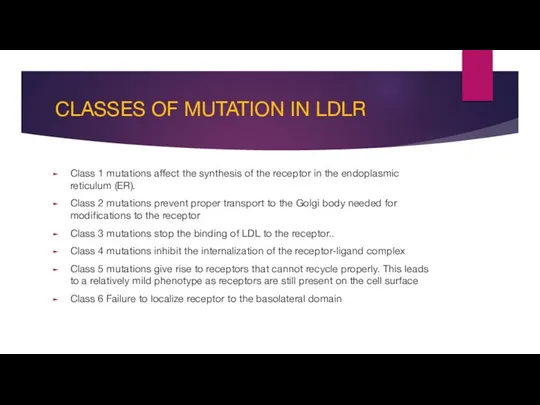
- 10. CLASSES OF MUTATION IN LDLR Class 1 mutations affect the synthesis of the receptor in the
- 11. Mutation in APOE gene At least five mutations in the APOB gene are known to cause
- 12. Function of LDLRAP1 Gene The LDLRAP1 gene is located on 1p36-p35. The LDLRAP1 gene is also
- 13. Mutation in LDLRAP1 gene More than 10 mutations in the LDLRAP1 gene have been shown to
- 14. FUNCTION OF PCSK9 GENE The PCSK9 protein appears to control the number of low-density lipoprotein receptors,
- 15. TREATMENT Heterozygous FH is normally treated with statins-drugs that lower cholesterol level Bile acid sequestrants (hypolipidemic
- 17. Скачать презентацию
Слайд 2Introduction
Familial hypercholesterolemia (FH) have raised cholesterol levels in blood with a
Introduction
Familial hypercholesterolemia (FH) have raised cholesterol levels in blood with a

Слайд 3CLINICAL MANIFESTATIONS
High cholesterol level in blood.
Heterozygotes may have premature cardiovascular disease
CLINICAL MANIFESTATIONS
High cholesterol level in blood.
Heterozygotes may have premature cardiovascular disease

Слайд 4A- Xanthelasma
B – Corneal arcus (Arcus
senilis)
C - Achilles tendon
A- Xanthelasma
B – Corneal arcus (Arcus
senilis)
C - Achilles tendon
Слайд 5PLASMA CHOLESTEROL LEVEL IN NORMAL AND FH INDIVIDUALS
NORMAL – 150 – 200
PLASMA CHOLESTEROL LEVEL IN NORMAL AND FH INDIVIDUALS
NORMAL – 150 – 200

Слайд 6Function of LDLR gene
The LDLR gene provides instructions for making a protein
Function of LDLR gene
The LDLR gene provides instructions for making a protein

Слайд 7Mutation in LDLR gene
Mutations in the LDLR gene cause FH
More than
Mutation in LDLR gene
Mutations in the LDLR gene cause FH
More than

Слайд 10CLASSES OF MUTATION IN LDLR
Class 1 mutations affect the synthesis of the
CLASSES OF MUTATION IN LDLR
Class 1 mutations affect the synthesis of the
Слайд 11Mutation in APOE gene
At least five mutations in the APOB gene are
Mutation in APOE gene
At least five mutations in the APOB gene are

Слайд 12Function of LDLRAP1 Gene
The LDLRAP1 gene is located on 1p36-p35.
The LDLRAP1
Function of LDLRAP1 Gene
The LDLRAP1 gene is located on 1p36-p35.
The LDLRAP1

Слайд 13Mutation in LDLRAP1 gene
More than 10 mutations in the LDLRAP1 gene have
Mutation in LDLRAP1 gene
More than 10 mutations in the LDLRAP1 gene have

Слайд 14FUNCTION OF PCSK9 GENE
The PCSK9 protein appears to control the number of
FUNCTION OF PCSK9 GENE
The PCSK9 protein appears to control the number of

Слайд 15 TREATMENT
Heterozygous FH is normally treated with statins-drugs that lower cholesterol level
TREATMENT
Heterozygous FH is normally treated with statins-drugs that lower cholesterol level

 Animals vocabulary review by herber
Animals vocabulary review by herber Методы решения текстовых задач
Методы решения текстовых задач Социальные функции и социальный механизм действия права
Социальные функции и социальный механизм действия права Понятие, признаки и структура норм права
Понятие, признаки и структура норм права На пути к жизненному успеху
На пути к жизненному успеху Ацетилен и его гомологи
Ацетилен и его гомологи die Schweiz
die Schweiz Классы решений для финансового сектора
Классы решений для финансового сектора Отчёт учителя английского языка Полуэктовой Е.С. за март- июнь 2012г.
Отчёт учителя английского языка Полуэктовой Е.С. за март- июнь 2012г. 40 ГОДИНИ СПЕЦИАЛНОСТ “ИНФОРМАТИКА” Ние сме във времето и времето е в нас... [Васил Левски]
40 ГОДИНИ СПЕЦИАЛНОСТ “ИНФОРМАТИКА” Ние сме във времето и времето е в нас... [Васил Левски] Презентация на тему Модификационная изменчивость 9 - 10 класс
Презентация на тему Модификационная изменчивость 9 - 10 класс Великие ученые
Великие ученые ГОУ ВПО ХМАО-Югры «Ханты-Мансийская государственная медицинская академия»
ГОУ ВПО ХМАО-Югры «Ханты-Мансийская государственная медицинская академия» Статистика рынка Казахстана с точки зрения карточных интернет транзакций
Статистика рынка Казахстана с точки зрения карточных интернет транзакций Щи
Щи Продвижение в социальных сетях и реклама в блогах
Продвижение в социальных сетях и реклама в блогах Отчет о ходе реализации целевой программы Калининградской области «Развитие здравоохранения Калининградской области на период
Отчет о ходе реализации целевой программы Калининградской области «Развитие здравоохранения Калининградской области на период  Презентация на тему Ферменты
Презентация на тему Ферменты Что мы знаем о местоимении?
Что мы знаем о местоимении? Работы в технике айрисфолдинг
Работы в технике айрисфолдинг Психосексуальное развитие детей и подростков
Психосексуальное развитие детей и подростков О ФОРМИРОВАНИИ АРХИВНОГО ФОНДА ДАННЫХ О СОСТОЯНИИ ОКРУЖАЮЩЕЙ СРЕДЫ, ЕЕ ЗАГРЯЗНЕНИИ
О ФОРМИРОВАНИИ АРХИВНОГО ФОНДА ДАННЫХ О СОСТОЯНИИ ОКРУЖАЮЩЕЙ СРЕДЫ, ЕЕ ЗАГРЯЗНЕНИИ «Социальная реабилитация людей с особенностями психофизического развития и организация временной занятости молодежи».
«Социальная реабилитация людей с особенностями психофизического развития и организация временной занятости молодежи». Тема:
Тема: Создание индивидуального стиля заказчика по мотивам ретро-стилей 50-70 годов ХХ века
Создание индивидуального стиля заказчика по мотивам ретро-стилей 50-70 годов ХХ века Надежность технологической системы
Надежность технологической системы Bed_Wars
Bed_Wars Эрнест Хемингуэй. Старик и море
Эрнест Хемингуэй. Старик и море