- КОМП’ЮТЕРНЕ МОДЕЛЮВАННЯ ЛІКАРСЬКИХ ЗАСОБІВ: КВАНТОВО-ФАРМАКОЛОГІЧНИЙ АСПЕКТ

Содержание
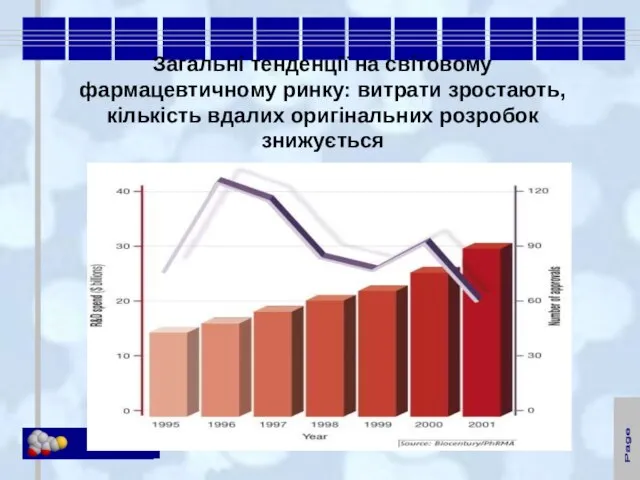
- 2. Загальні тенденції на світовому фармацевтичному ринку: витрати зростають, кількість вдалих оригінальних розробок знижується
- 3. 1 Розробка оригінального лікарського засобу – унікальний мультидисциплінарний процес, спрямований на створення нового терапевтичного агента з
- 4. 2 Причини невдач, з якими стикається дослідник в процесі впровадження в медичну практику оригінальних ліків: недосконалі
- 5. Традиційний шлях розробки оригінального препарата Ідентифікація хвороби Виділення білка, пов’язаного з хворобою (2-5 років) Пошук ефективного
- 6. Впровадження сучасних технологій в розробку ліків За останні декілька років з’явилися «революційні» технології : Генні чипи,
- 7. Ідентифікація хвороби Геноміка, протеоміка Високоефективний скринінг Молекулярне моделювання Віртуальный скринінг Комбінаторна хімія IN VITRO та IN
- 10. Хемоінформатика – комп’ютерне передбачення біологічної активності речовин
- 11. 3 З’ясування залежності між хімічною будовою речовини, їх фізико-хімічними властивостями та біологічною активністю (Quantitative Structure-Activity Relationship)
- 12. 4 Найбільш значні успіхи природничих наук, що дали змогу досліджувати структуру біомолекул: вдосконалення рентгеноструктурного аналізу –
- 13. 5 Принципові завдання комп’ютерного моделювання: розрахункове відтворення (побудова) різноманітних систем та їх властивостей; розрахункове відтворення різноманітних
- 14. 6 Використання комп’ютерного моделювання в фізико-хімічній фармакології: розрахунки будови і спектрів молекул та інших атомно-молекулярних систем
- 15. Квантова фармакологія: - розділ науки, в якому знання електронної структури препаратів використовується для de novo дизайна
- 16. 7 Програми з розрахунків квантово-хімічних властивостей використовуються в сучасній фармакології для вирішення наступних завдань: встановлення структури
- 17. 8 HyperChem - програма для комп’ютерного моделювання молекул та дослідження квантово- хімічних параметрів молекулярної динаміки. До
- 18. 9 Квантово-хімічний комплекс GAMESS: дозволяє проводити розрахунки шляхом врахування параметрів ab intio може бути інтегрований у
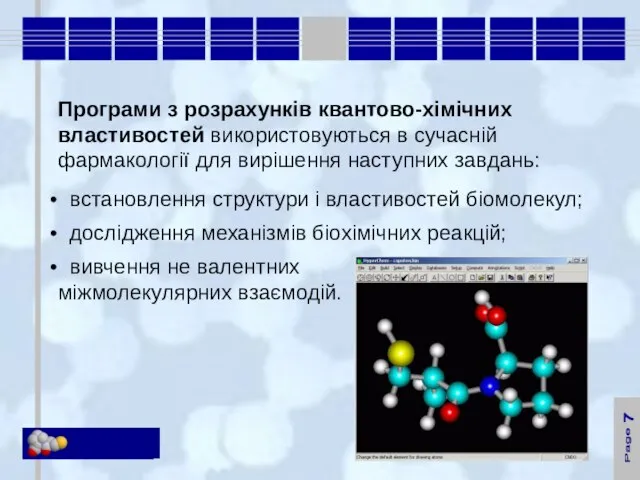
- 19. 10 Дослідження квантово-хімічних властивостей селективного α1-адреноблокатора празозина
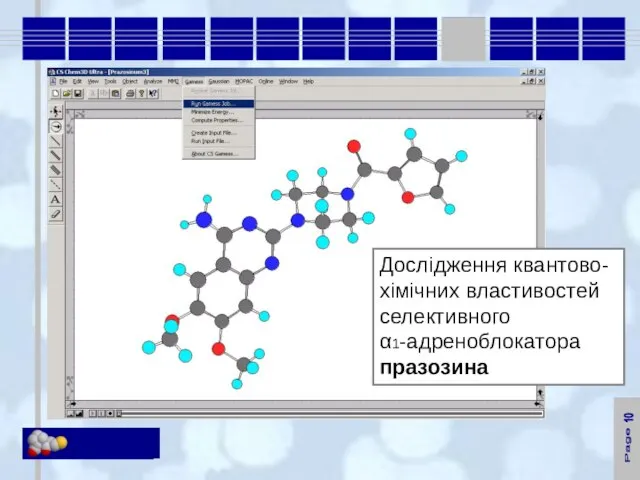
- 20. 11 Заряди на атомах та напрямок диполю в молекулі празозина
- 21. 12 Локалізація вищої зайнятої та нижчої вільної молекулярних орбіталей в молекулі празозина
- 22. 13 Розподіл електростатичного потенціалу в молекулі празозина
- 23. 14 Енергетичні властивості молекули празозину Number of electrons = 146 Number of Double Occupied Levels =
- 24. 15 Висновки Програма GAMESS поступається програмі HyperChem за такими показниками, як спектр виконуваних задач та адаптованість
- 25. 16 Перспективи: Розрахунки квантово-хімічних параметрів взаємодії празозину з біолігандами у водному розчині та моделювання взаємодії празозину
- 27. Скачать презентацию
Слайд 2Загальні тенденції на світовому фармацевтичному ринку: витрати зростають, кількість вдалих оригінальних розробок
Загальні тенденції на світовому фармацевтичному ринку: витрати зростають, кількість вдалих оригінальних розробок
Слайд 31
Розробка оригінального лікарського засобу – унікальний мультидисциплінарний процес, спрямований на створення нового
1
Розробка оригінального лікарського засобу – унікальний мультидисциплінарний процес, спрямований на створення нового

Слайд 42
Причини невдач, з якими стикається дослідник в процесі впровадження в медичну практику
2
Причини невдач, з якими стикається дослідник в процесі впровадження в медичну практику

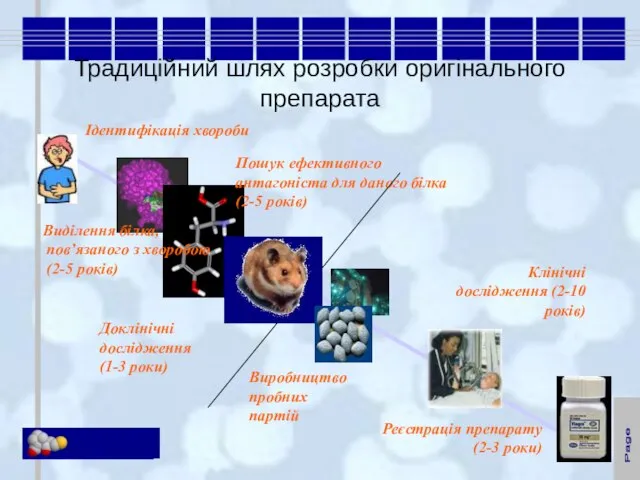
Слайд 5Традиційний шлях розробки оригінального препарата
Ідентифікація хвороби
Виділення білка,
пов’язаного з хворобою
(2-5 років)
Пошук
Традиційний шлях розробки оригінального препарата
Ідентифікація хвороби
Виділення білка,
пов’язаного з хворобою
(2-5 років)
Пошук
Слайд 6Впровадження сучасних технологій в розробку ліків
За останні декілька років з’явилися «революційні» технології
Впровадження сучасних технологій в розробку ліків
За останні декілька років з’явилися «революційні» технології

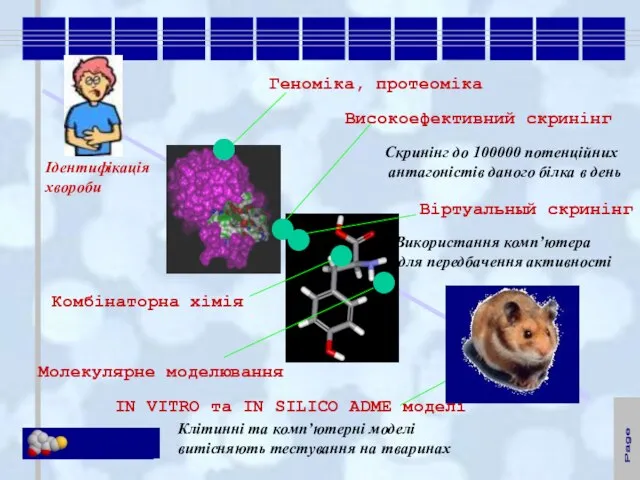
Слайд 7Ідентифікація
хвороби
Геноміка, протеоміка
Високоефективний скринінг
Молекулярне моделювання
Віртуальный скринінг
Комбінаторна хімія
IN VITRO та IN SILICO ADME
Ідентифікація
хвороби
Геноміка, протеоміка
Високоефективний скринінг
Молекулярне моделювання
Віртуальный скринінг
Комбінаторна хімія
IN VITRO та IN SILICO ADME


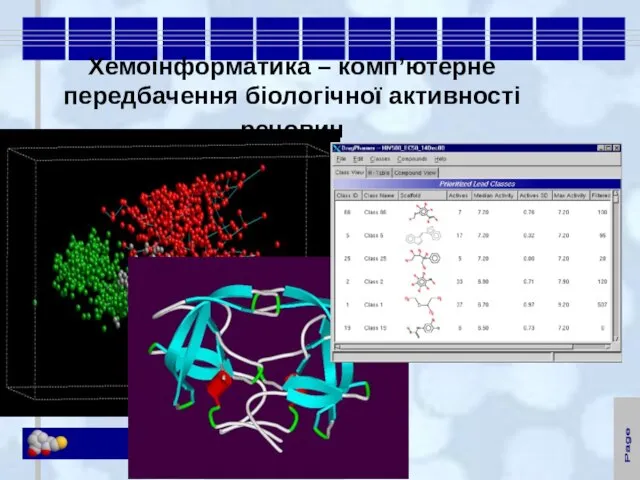
Слайд 10Хемоінформатика – комп’ютерне передбачення біологічної активності речовин
Хемоінформатика – комп’ютерне передбачення біологічної активності речовин
Слайд 113
З’ясування залежності між хімічною будовою речовини, їх фізико-хімічними властивостями та біологічною активністю
3
З’ясування залежності між хімічною будовою речовини, їх фізико-хімічними властивостями та біологічною активністю
Слайд 124
Найбільш значні успіхи природничих наук, що дали змогу досліджувати структуру біомолекул:
4
Найбільш значні успіхи природничих наук, що дали змогу досліджувати структуру біомолекул:

Слайд 135
Принципові завдання комп’ютерного моделювання:
розрахункове відтворення (побудова) різноманітних систем та їх властивостей;
5
Принципові завдання комп’ютерного моделювання:
розрахункове відтворення (побудова) різноманітних систем та їх властивостей;

Слайд 146
Використання комп’ютерного моделювання в фізико-хімічній фармакології:
розрахунки будови і спектрів молекул та
6
Використання комп’ютерного моделювання в фізико-хімічній фармакології:
розрахунки будови і спектрів молекул та

Слайд 15Квантова фармакологія:
- розділ науки, в якому знання електронної структури препаратів використовується для
Квантова фармакологія:
- розділ науки, в якому знання електронної структури препаратів використовується для

Слайд 167
Програми з розрахунків квантово-хімічних властивостей використовуються в сучасній фармакології для вирішення наступних
7
Програми з розрахунків квантово-хімічних властивостей використовуються в сучасній фармакології для вирішення наступних
Слайд 178
HyperChem - програма для комп’ютерного моделювання молекул та дослідження квантово- хімічних параметрів молекулярної
8
HyperChem - програма для комп’ютерного моделювання молекул та дослідження квантово- хімічних параметрів молекулярної

Слайд 189
Квантово-хімічний комплекс GAMESS:
дозволяє проводити розрахунки шляхом врахування параметрів ab intio
9
Квантово-хімічний комплекс GAMESS:
дозволяє проводити розрахунки шляхом врахування параметрів ab intio

Слайд 1910
Дослідження квантово-хімічних властивостей селективного
α1-адреноблокатора празозина
10
Дослідження квантово-хімічних властивостей селективного
α1-адреноблокатора празозина
Слайд 2011
Заряди на атомах та напрямок диполю в молекулі празозина
11
Заряди на атомах та напрямок диполю в молекулі празозина
Слайд 2112
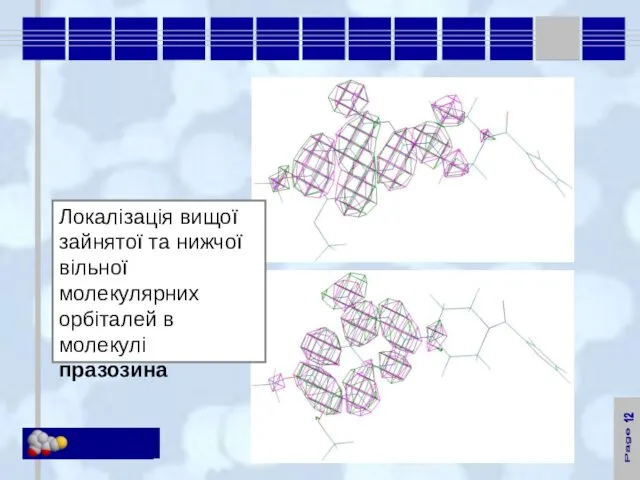
Локалізація вищої зайнятої та нижчої вільної молекулярних орбіталей в молекулі празозина
12
Локалізація вищої зайнятої та нижчої вільної молекулярних орбіталей в молекулі празозина
Слайд 2213
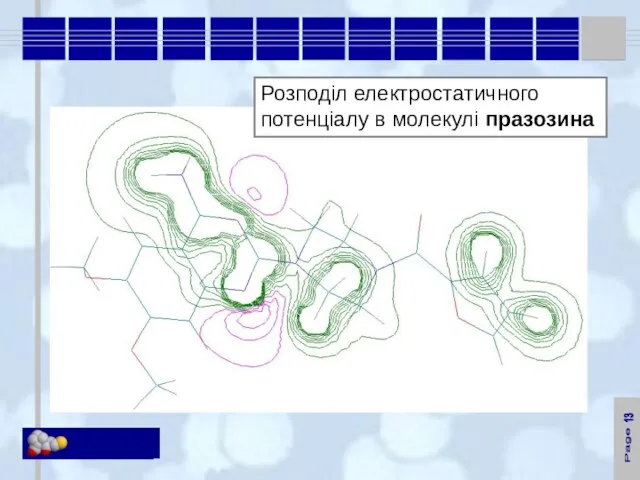
Розподіл електростатичного потенціалу в молекулі празозина
13
Розподіл електростатичного потенціалу в молекулі празозина
Слайд 2314
Енергетичні властивості
молекули празозину
Number of electrons = 146
Number of Double Occupied Levels
14
Енергетичні властивості
молекули празозину
Number of electrons = 146
Number of Double Occupied Levels

Слайд 2415
Висновки
Програма GAMESS поступається програмі HyperChem за такими показниками, як спектр виконуваних
15
Висновки
Програма GAMESS поступається програмі HyperChem за такими показниками, як спектр виконуваних

Слайд 2516
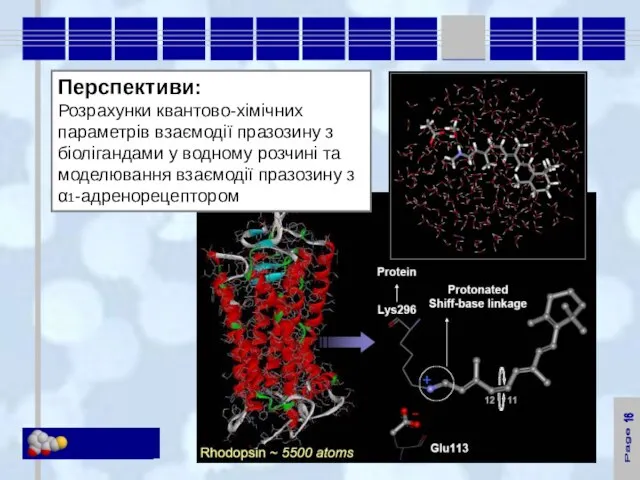
Перспективи:
Розрахунки квантово-хімічних параметрів взаємодії празозину з біолігандами у водному розчині та моделювання
16
Перспективи:
Розрахунки квантово-хімічних параметрів взаємодії празозину з біолігандами у водному розчині та моделювання
 «НАША НОВАЯ ШКОЛА» Послание Президента РФ Д.А.Медведева Федеральному собранию Российской Федерации 5 ноября 2008г.
«НАША НОВАЯ ШКОЛА» Послание Президента РФ Д.А.Медведева Федеральному собранию Российской Федерации 5 ноября 2008г. Введение в дизайн. Основные понятия
Введение в дизайн. Основные понятия Транспорт. Спецтехника
Транспорт. Спецтехника КЛИЕНТОРИЕНТИРОВАННЫЕ ОБРАЗОВАТЕЛЬНЫЕ ОРГАНИЗАЦИИ
КЛИЕНТОРИЕНТИРОВАННЫЕ ОБРАЗОВАТЕЛЬНЫЕ ОРГАНИЗАЦИИ Международные отношения на американском континенте. Панамериканские конференции 1920-х годов
Международные отношения на американском континенте. Панамериканские конференции 1920-х годов Биполярные транзисторы (лекция 4)
Биполярные транзисторы (лекция 4) ИнфраФонд РВК
ИнфраФонд РВК ИНТЕГРИРОВАННАЯ СИСТЕМА УПРАВЛЕНИЯ СТРОИТЕЛЬНОЙ КОМПАНИЕЙ
ИНТЕГРИРОВАННАЯ СИСТЕМА УПРАВЛЕНИЯ СТРОИТЕЛЬНОЙ КОМПАНИЕЙ Архітектурні пам’ятки Городенківщини
Архітектурні пам’ятки Городенківщини Модерн в Казани
Модерн в Казани Сирень
Сирень Есть контакт
Есть контакт Осцилляторы. Импульсные возбудители дуги. Балластные реостаты
Осцилляторы. Импульсные возбудители дуги. Балластные реостаты Энергия солнца в Вашем Доме!
Энергия солнца в Вашем Доме! Правописание безударных гласных в корне слова
Правописание безударных гласных в корне слова Sketch Meme
Sketch Meme Lomonosov
Lomonosov Муниципальное общеобразовательное учреждениеЦентр образования города Тулуна
Муниципальное общеобразовательное учреждениеЦентр образования города Тулуна Права потребителей: общие и специальные
Права потребителей: общие и специальные Аландское городище
Аландское городище Государственно-общественное управление образованием в условиях реализации №83-ФЗ
Государственно-общественное управление образованием в условиях реализации №83-ФЗ Гибкость и ловкость. Круговой метод тренировки для развития основных групп мышц
Гибкость и ловкость. Круговой метод тренировки для развития основных групп мышц Обмен липидов-3
Обмен липидов-3 Отчет о результатах социологического исследования "Киев - районные выборы"
Отчет о результатах социологического исследования "Киев - районные выборы" Тематика, основные цели, задачи, результаты муниципальных семинаров
Тематика, основные цели, задачи, результаты муниципальных семинаров Экспонир. устр-во с зап. на внешн. поверхн. барабана
Экспонир. устр-во с зап. на внешн. поверхн. барабана Линейная Автоматика
Линейная Автоматика Снабжение и логистика на предприятии
Снабжение и логистика на предприятии