- Первичные иммунодефициты (ПИД)

Содержание
- 2. Регистр пациентов с ПИД В Российском регистре более 1700 пациентов. Наиболее часто выявляется селективный дефицит IgA
- 3. Классификация 2007 года 8 групп ПИД С учетом уровня нарушения, преимущественного дефицита и преимущественной клинической картины
- 4. Первичные иммунодефициты - классификация 1.Связанные с дефектами фагоцитов 2.Дефицит системы комплемента 3. Т-клеточные иммунодефициты 4. В-клеточные
- 5. ПИД с поражением фагоцитарного звена Отмечаются такие дефекты как нарушение подвижности, хемотаксиса, адгезии, эндоцитоза, киллинга, секреции
- 6. ПИД с поражением фагоцитарного звена Хронический гранулематоз Синдром Чегиак-Хигаши (Чедиак-Хигаси)
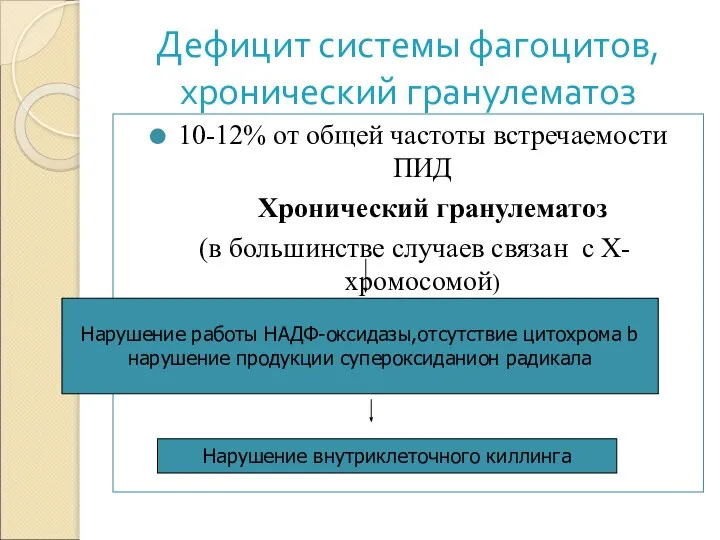
- 7. Дефицит системы фагоцитов, хронический гранулематоз 10-12% от общей частоты встречаемости ПИД Хронический гранулематоз (в большинстве случаев
- 8. Клинические проявления хронического гранулематоза Рецидивирующие бактериальные и грибковые инфекции (органы дыхания, кожа, лимфатические узлы). Лимфаденит, гепатоспленомегалия.
- 9. Синдром Чегиак-Хигаши (Чедиак-Хигаси) Потеря нейтрофилами способности высвобождать лизосомальные ферменты Нарушение хемотаксиса Нарушение противоопухолевого иммунитета
- 10. Клинические проявления Пиогенные рецидивирующие инфекции Альбинизм (высокая фоточувствительность кожи) Средний возраст больных- 6-7 лет, смерть от
- 11. Дефицит системы комплемента 1% от общего количества перв.ИД Генетические дефекты описаны для всех компонентов комплемента С1q
- 12. Т-клеточные ИД Синдром Ди Джорджи Синдром Незелофа Хронический слизисто-кожный кандидоз
- 13. Т-клеточный ИД, Синдром Ди-Джорджи (синдром делеции 22 хромосомы) порок развития первого и второго жаберных карманов –
- 14. Синдром Ди Джорджи Триада -недоразвитие тимуса -отсутствие паращитовидных желез -аномалия развития сердечно-сосудистой системы Классификация синдрома Ди
- 15. Синдром Ди Джорджи Иммунологические последствия Снижение количества и активности Т-лф Количество В-лф в норме Уровень ат
- 16. Синдром Ди Джорджи Клиника: Рецидивирующие вирусные, паразитарные, вирусные инфекции Судороги (полный Ди Джоржди) Дисморфия лица –
- 17. Больная с синдромом Ди-Джорджи 11 лет ОРВИ 4-5 раз в год 10 лет –аутоиммунная тромбоцитопеническая пурпура
- 18. Синдром Незелофа (лимфоцитарная дисгенезия, нормоплазмоцитарная и нормогаммаглобулинемическая аплазия) Гипоплазия тимуса АТ – в норме
- 19. Синдром Незелофа клиника: замедление роста рецидивирующие инфекции кожи и легких кандидоз лимфаденопатия
- 20. Хронический слизисто-кожный кандидоз Селективный дефицит ответа Т-клеток на Аг Candida albicans Ответ на другие аг может
- 21. Хронический кандидоз слизистых и кожи
- 22. Дефицит В-клеточного звена Составляет 50-70% общего количества ПИД Как правило, протекают в более легкой форме по
- 23. Дефицит В-клеточного звена болезнь Брутона Агамма (гипогамма) глобулинемия (болезнь Брутона) рецессивный тип наследования, Х-сцепленный Дефицит цитоплазматической
- 24. Клинические проявления Определяется на 5-9 месяце жизни (когда материнские АТ перестают защищать организм ребенка) Рецидивирующие гнойные
- 25. Иммунологические признаки Очень низкие уровни Ig всех классов Отсутствие циркулирующих В лимфоцитов Гипоплазия миндалин Отсутствие плазматических
- 26. ТКИД Швейцарский тип Синдром Вискотта-Олдрича )сцепленный с Х хромосомой) Атаксия-телеангиэктазия (Синдром Луи-Бара) Сцепленный с Х-хромосомой ИД
- 27. ТКИД – основные клинические особенности • Раннее начало (обычно между 3-9 месяцами жизни) • Гипоплазия лимфоидной
- 28. ТКИД (швейцарский тип) Х-сцепленный тип Дефект на уровне стволовой клетки нежизнеспособность
- 29. Синдром Вискотта-Олдрича (СВО) - ТКИД В основе лежит мутация гена, который кодирует белок (WASP), отвечающий за
- 30. Комбинированные ИД Синдром Вискотта-Олдрича Нарушение активации Т-h и Т-s ) Врожденный дефект тромбоцитов Нарушение способности макрофагов
- 31. Синдром Вискотта-Олдрича Клиника: Экзема Тромбоцитопения Бактериальные и вирусные инфекции
- 32. Больной с СВО 4-х лет • С 3 месяцев до 3-х лет Тяжелый распространенный атопический дерматит
- 33. Синдром Вискотта-Олдрича
- 34. Синдром Луи-Бар (атаксия-телеангиэктазия) Гипоплазия тимуса Гипоплазия лимф. узлов, селезенки Количественная и функциональная недостаточность Т-лимфоцитов Снижение количества
- 35. Синдром Луи-Бар Клиника: Атаксия Телеангиэктазы (на носу, ушах, конъюктиве) могут появляться в возрасте от 2 до
- 36. Синдром Луи-Бар
- 37. Где ошибки?
- 39. ВТОРИЧНЫЕ ИД ВИД – клинико-иммунологический синдром. Особенности: 1. нарушения иммунитета вторичны (изначально иммунная система нормально сформирована
- 40. Классификация ВИД: По темпам развития: острый ИД, хронический ИД. По уровню нарушения: Т-клеточный, В-клеточный, нарушение фагоцитов,
- 41. Причины развития ВИД: 1. Инфекции: вирусные, бактериальные, протозойные. 2. Нарушения питания. 3.Злокачественные новообразования. 4. ИД после
- 42. ИД, вызванные цитомегаловирусом Цитомегаловирус человека (Cytomegalovirus hominis, Вирус Герпеса Человека 5 типа). Краткое название: ЦМВ, ВГЧ-5,
- 43. Строение цитомегаловируса Сферической формы Геном представлен 2-нитевой ДНК Тип симметрии кубический (р65,р70) Суперкапсид gp 116, gp58,
- 44. Патогенез Источником инфекции является больной человек или вирусоноситель. Заражение происходит воздушно-капельным, контактным, пищевым, парентеральным, траспланцентарным путем.
- 45. Иммунопатогенез Репликация вируса происходит в лейкоцитах, клетках системы мононуклеарных фагоцитов. Процесс репликации заканчивается формированием дочерних вирусных
- 47. Клинические формы Скрытая форма Субклиническая форма Манифестная форма Со временем – нарастание ИД состояния
- 48. Вирус Эпштейна-Барра Вирус Эпштейна-Барр (Epstein-Barr virus - EBV), так же называемый человеческим герпесвирусом вирусом 4 (Human
- 49. Вирус Эпштейна—Барра Вирус содержит ДНК; вирион состоит из капсида диаметром 120—150 нм, окруженного оболочкой, содержащей липиды.
- 50. Патогенез Источник инфекции — больной человек, в том числе и больные стертыми формами болезни. Передача инфекции
- 52. Скачать презентацию
Слайд 2Регистр пациентов с ПИД
В Российском регистре более 1700 пациентов.
Наиболее часто выявляется селективный
Регистр пациентов с ПИД
В Российском регистре более 1700 пациентов.
Наиболее часто выявляется селективный

Слайд 3Классификация 2007 года
8 групп ПИД
С учетом уровня нарушения, преимущественного дефицита и
Классификация 2007 года
8 групп ПИД
С учетом уровня нарушения, преимущественного дефицита и

Слайд 4Первичные иммунодефициты - классификация
1.Связанные с дефектами фагоцитов
2.Дефицит системы комплемента
3. Т-клеточные иммунодефициты
4.
Первичные иммунодефициты - классификация
1.Связанные с дефектами фагоцитов
2.Дефицит системы комплемента
3. Т-клеточные иммунодефициты
4.

Слайд 5ПИД с поражением фагоцитарного звена
Отмечаются такие дефекты как нарушение подвижности, хемотаксиса, адгезии,
ПИД с поражением фагоцитарного звена
Отмечаются такие дефекты как нарушение подвижности, хемотаксиса, адгезии,

Слайд 6ПИД с поражением фагоцитарного звена
Хронический гранулематоз
Синдром Чегиак-Хигаши (Чедиак-Хигаси)
ПИД с поражением фагоцитарного звена
Хронический гранулематоз
Синдром Чегиак-Хигаши (Чедиак-Хигаси)

Слайд 7Дефицит системы фагоцитов,
хронический гранулематоз
10-12% от общей частоты встречаемости ПИД
Хронический гранулематоз
(в
Дефицит системы фагоцитов,
хронический гранулематоз
10-12% от общей частоты встречаемости ПИД
Хронический гранулематоз
(в
Слайд 8Клинические проявления хронического гранулематоза
Рецидивирующие бактериальные и грибковые инфекции (органы дыхания, кожа, лимфатические
Клинические проявления хронического гранулематоза
Рецидивирующие бактериальные и грибковые инфекции (органы дыхания, кожа, лимфатические

Слайд 9Синдром Чегиак-Хигаши
(Чедиак-Хигаси)
Потеря нейтрофилами способности высвобождать лизосомальные ферменты
Нарушение хемотаксиса
Нарушение противоопухолевого иммунитета
Синдром Чегиак-Хигаши
(Чедиак-Хигаси)
Потеря нейтрофилами способности высвобождать лизосомальные ферменты
Нарушение хемотаксиса
Нарушение противоопухолевого иммунитета

Слайд 10Клинические проявления
Пиогенные рецидивирующие инфекции
Альбинизм (высокая фоточувствительность кожи)
Средний возраст больных- 6-7 лет, смерть
Клинические проявления
Пиогенные рецидивирующие инфекции
Альбинизм (высокая фоточувствительность кожи)
Средний возраст больных- 6-7 лет, смерть

Слайд 11Дефицит системы комплемента
1% от общего количества перв.ИД
Генетические дефекты описаны для всех компонентов
Дефицит системы комплемента
1% от общего количества перв.ИД
Генетические дефекты описаны для всех компонентов

Слайд 12Т-клеточные ИД
Синдром Ди Джорджи
Синдром Незелофа
Хронический слизисто-кожный кандидоз
Т-клеточные ИД
Синдром Ди Джорджи
Синдром Незелофа
Хронический слизисто-кожный кандидоз

Слайд 13
Т-клеточный ИД,
Синдром Ди-Джорджи
(синдром делеции 22 хромосомы)
порок развития первого и второго жаберных
карманов
Т-клеточный ИД,
Синдром Ди-Джорджи
(синдром делеции 22 хромосомы)
порок развития первого и второго жаберных
карманов

Слайд 14Синдром Ди Джорджи
Триада
-недоразвитие тимуса
-отсутствие паращитовидных желез
-аномалия развития сердечно-сосудистой
Синдром Ди Джорджи
Триада
-недоразвитие тимуса
-отсутствие паращитовидных желез
-аномалия развития сердечно-сосудистой

Слайд 15Синдром Ди Джорджи
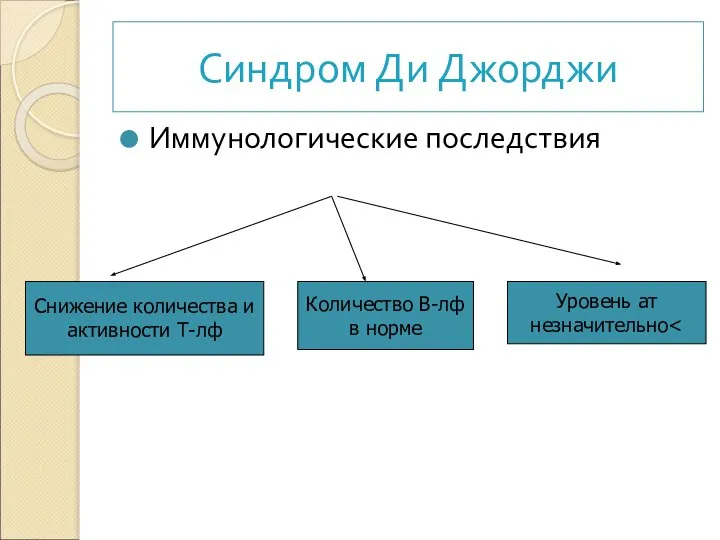
Иммунологические последствия
Снижение количества и
активности Т-лф
Количество В-лф
в норме
Уровень ат
Синдром Ди Джорджи
Иммунологические последствия
Снижение количества и
активности Т-лф
Количество В-лф
в норме
Уровень ат
Слайд 16Синдром
Ди Джорджи
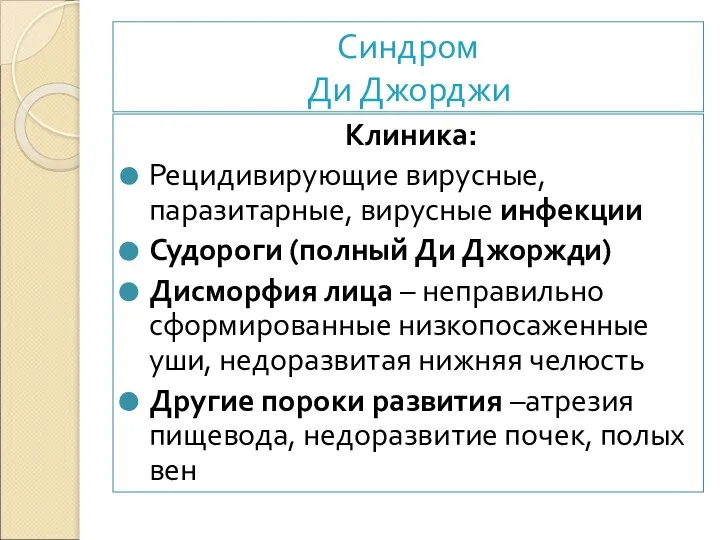
Клиника:
Рецидивирующие вирусные, паразитарные, вирусные инфекции
Судороги (полный Ди Джоржди)
Дисморфия лица –
Синдром
Ди Джорджи
Клиника:
Рецидивирующие вирусные, паразитарные, вирусные инфекции
Судороги (полный Ди Джоржди)
Дисморфия лица –
Слайд 17Больная с синдромом
Ди-Джорджи 11 лет
ОРВИ 4-5 раз в год
10 лет –аутоиммунная
тромбоцитопеническая пурпура
Больная с синдромом
Ди-Джорджи 11 лет
ОРВИ 4-5 раз в год
10 лет –аутоиммунная
тромбоцитопеническая пурпура

Слайд 18Синдром Незелофа (лимфоцитарная дисгенезия, нормоплазмоцитарная и нормогаммаглобулинемическая аплазия)
Гипоплазия тимуса
АТ –
Синдром Незелофа (лимфоцитарная дисгенезия, нормоплазмоцитарная и нормогаммаглобулинемическая аплазия)
Гипоплазия тимуса
АТ –

Слайд 19Синдром Незелофа
клиника:
замедление роста
рецидивирующие инфекции кожи и легких
кандидоз
лимфаденопатия
Синдром Незелофа
клиника:
замедление роста
рецидивирующие инфекции кожи и легких
кандидоз
лимфаденопатия

Слайд 20Хронический
слизисто-кожный кандидоз
Селективный дефицит ответа Т-клеток на Аг Candida albicans
Ответ на другие
Хронический
слизисто-кожный кандидоз
Селективный дефицит ответа Т-клеток на Аг Candida albicans
Ответ на другие

Слайд 21Хронический кандидоз
слизистых и кожи
Хронический кандидоз
слизистых и кожи
Слайд 22Дефицит В-клеточного звена
Составляет 50-70% общего количества ПИД
Как правило, протекают в более легкой
Дефицит В-клеточного звена
Составляет 50-70% общего количества ПИД
Как правило, протекают в более легкой

Слайд 23Дефицит В-клеточного звена
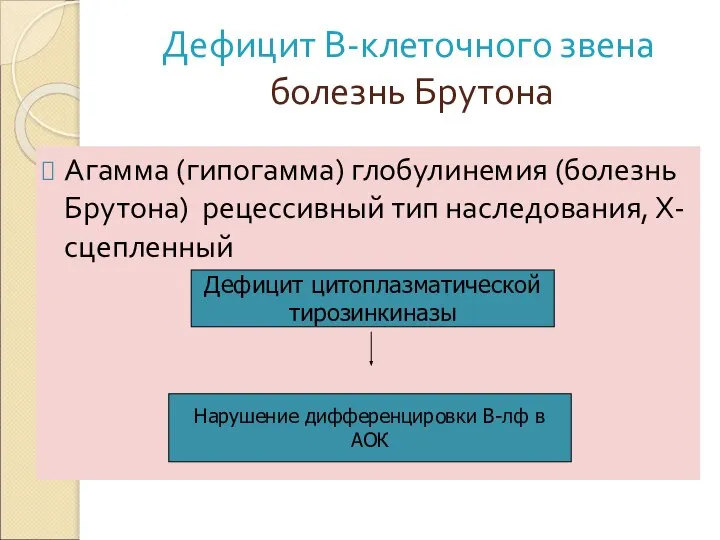
болезнь Брутона
Агамма (гипогамма) глобулинемия (болезнь Брутона) рецессивный тип наследования,
Дефицит В-клеточного звена
болезнь Брутона
Агамма (гипогамма) глобулинемия (болезнь Брутона) рецессивный тип наследования,
Слайд 24Клинические проявления
Определяется на 5-9 месяце жизни (когда материнские АТ перестают защищать организм
Клинические проявления
Определяется на 5-9 месяце жизни (когда материнские АТ перестают защищать организм

Слайд 25Иммунологические признаки
Очень низкие уровни Ig всех классов
Отсутствие циркулирующих В лимфоцитов
Гипоплазия миндалин
Отсутствие плазматических
Иммунологические признаки
Очень низкие уровни Ig всех классов
Отсутствие циркулирующих В лимфоцитов
Гипоплазия миндалин
Отсутствие плазматических

Слайд 26ТКИД
Швейцарский тип
Синдром Вискотта-Олдрича )сцепленный с Х хромосомой)
Атаксия-телеангиэктазия (Синдром Луи-Бара)
Сцепленный с Х-хромосомой ИД
ТКИД
Швейцарский тип
Синдром Вискотта-Олдрича )сцепленный с Х хромосомой)
Атаксия-телеангиэктазия (Синдром Луи-Бара)
Сцепленный с Х-хромосомой ИД

Слайд 27ТКИД – основные клинические особенности
• Раннее начало (обычно между 3-9 месяцами жизни)
•
ТКИД – основные клинические особенности
• Раннее начало (обычно между 3-9 месяцами жизни)
•

Слайд 28ТКИД (швейцарский тип)
Х-сцепленный тип
Дефект на уровне стволовой клетки
нежизнеспособность
ТКИД (швейцарский тип)
Х-сцепленный тип
Дефект на уровне стволовой клетки
нежизнеспособность

Слайд 29Синдром Вискотта-Олдрича
(СВО) - ТКИД
В основе лежит мутация гена,
который кодирует белок (WASP),
Синдром Вискотта-Олдрича
(СВО) - ТКИД
В основе лежит мутация гена,
который кодирует белок (WASP),

Слайд 30Комбинированные ИД
Синдром Вискотта-Олдрича
Нарушение активации Т-h и Т-s
<концентрация IgM, (IgA, IgE >)
Врожденный
Комбинированные ИД
Синдром Вискотта-Олдрича
Нарушение активации Т-h и Т-s
<концентрация IgM, (IgA, IgE >)
Врожденный

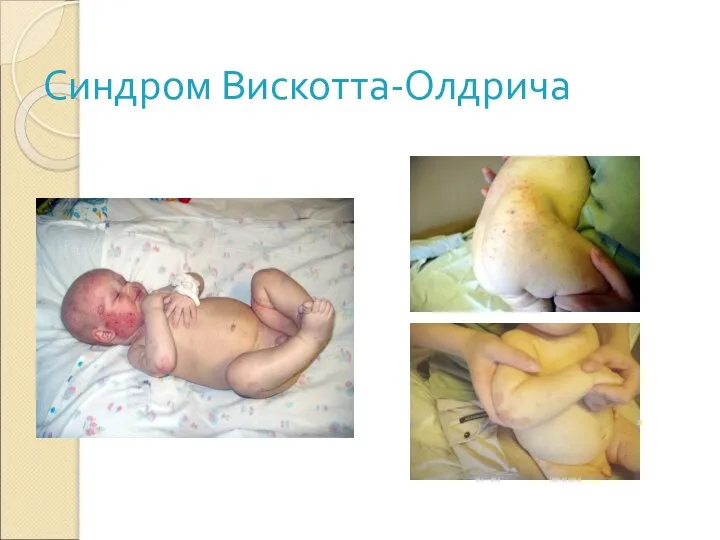
Слайд 31Синдром Вискотта-Олдрича
Клиника:
Экзема
Тромбоцитопения
Бактериальные и вирусные инфекции
Синдром Вискотта-Олдрича
Клиника:
Экзема
Тромбоцитопения
Бактериальные и вирусные инфекции

Слайд 32Больной с СВО 4-х лет
• С 3 месяцев до 3-х лет
Тяжелый распространенный
Больной с СВО 4-х лет
• С 3 месяцев до 3-х лет
Тяжелый распространенный

Слайд 33Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича
Слайд 34Синдром Луи-Бар
(атаксия-телеангиэктазия)
Гипоплазия тимуса
Гипоплазия лимф. узлов, селезенки
Количественная и функциональная недостаточность Т-лимфоцитов
Снижение количества IgA,
Синдром Луи-Бар
(атаксия-телеангиэктазия)
Гипоплазия тимуса
Гипоплазия лимф. узлов, селезенки
Количественная и функциональная недостаточность Т-лимфоцитов
Снижение количества IgA,

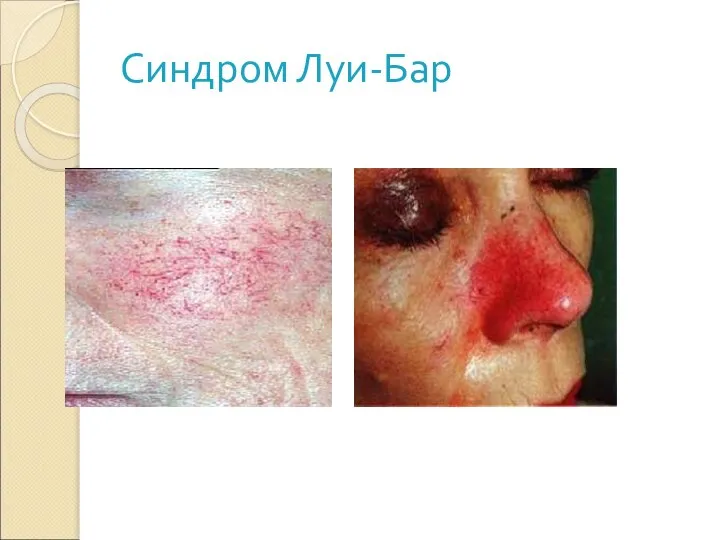
Слайд 35Синдром Луи-Бар
Клиника:
Атаксия
Телеангиэктазы
(на носу, ушах, конъюктиве) могут появляться в возрасте от 2
Синдром Луи-Бар
Клиника:
Атаксия
Телеангиэктазы
(на носу, ушах, конъюктиве) могут появляться в возрасте от 2

Слайд 36Синдром Луи-Бар
Синдром Луи-Бар
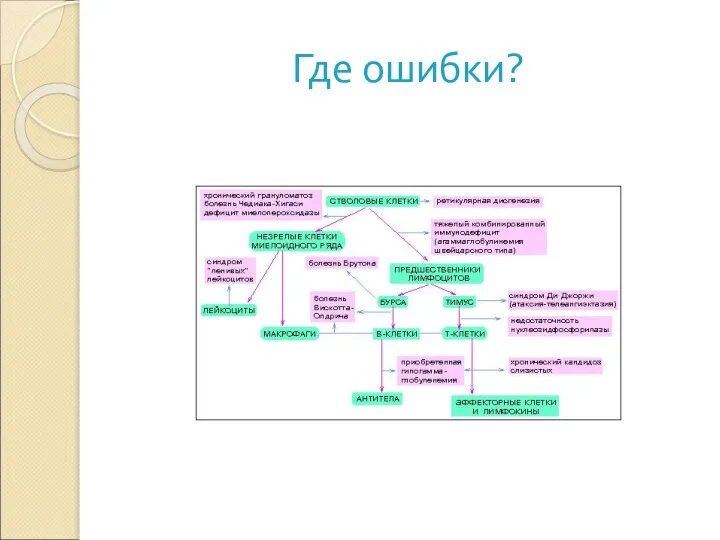
Слайд 37Где ошибки?
Где ошибки?

Слайд 39ВТОРИЧНЫЕ ИД
ВИД – клинико-иммунологический синдром.
Особенности:
1. нарушения иммунитета вторичны (изначально иммунная система
ВТОРИЧНЫЕ ИД
ВИД – клинико-иммунологический синдром.
Особенности:
1. нарушения иммунитета вторичны (изначально иммунная система

Слайд 40Классификация ВИД:
По темпам развития: острый ИД, хронический ИД.
По уровню нарушения: Т-клеточный, В-клеточный,
Классификация ВИД:
По темпам развития: острый ИД, хронический ИД.
По уровню нарушения: Т-клеточный, В-клеточный,

Слайд 41Причины развития ВИД:
1. Инфекции: вирусные, бактериальные, протозойные.
2. Нарушения питания.
3.Злокачественные новообразования.
4. ИД после
Причины развития ВИД:
1. Инфекции: вирусные, бактериальные, протозойные.
2. Нарушения питания.
3.Злокачественные новообразования.
4. ИД после

Слайд 42ИД, вызванные цитомегаловирусом
Цитомегаловирус человека (Cytomegalovirus hominis, Вирус Герпеса Человека 5 типа). Краткое
ИД, вызванные цитомегаловирусом
Цитомегаловирус человека (Cytomegalovirus hominis, Вирус Герпеса Человека 5 типа). Краткое

Слайд 43Строение цитомегаловируса
Сферической формы
Геном представлен 2-нитевой ДНК
Тип симметрии кубический (р65,р70)
Суперкапсид gp 116, gp58,
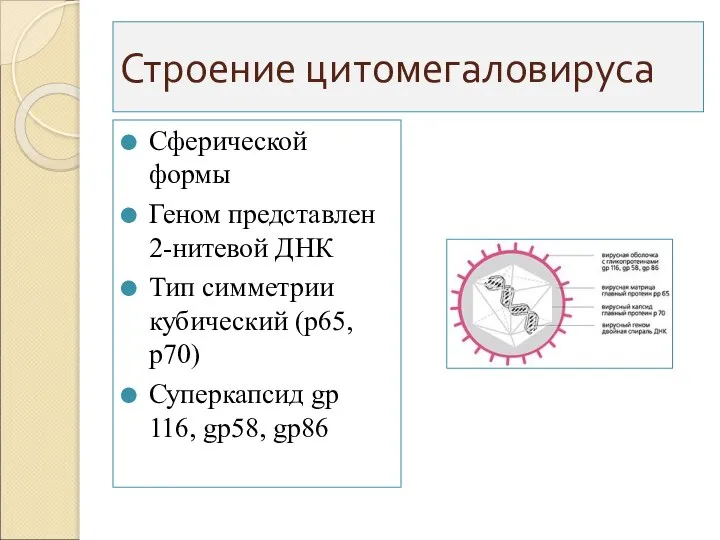
Строение цитомегаловируса
Сферической формы
Геном представлен 2-нитевой ДНК
Тип симметрии кубический (р65,р70)
Суперкапсид gp 116, gp58,
Слайд 44Патогенез
Источником инфекции является больной человек или вирусоноситель. Заражение происходит воздушно-капельным, контактным, пищевым,
Патогенез
Источником инфекции является больной человек или вирусоноситель. Заражение происходит воздушно-капельным, контактным, пищевым,

Слайд 45Иммунопатогенез
Репликация вируса происходит в лейкоцитах, клетках системы мононуклеарных фагоцитов.
Процесс репликации заканчивается
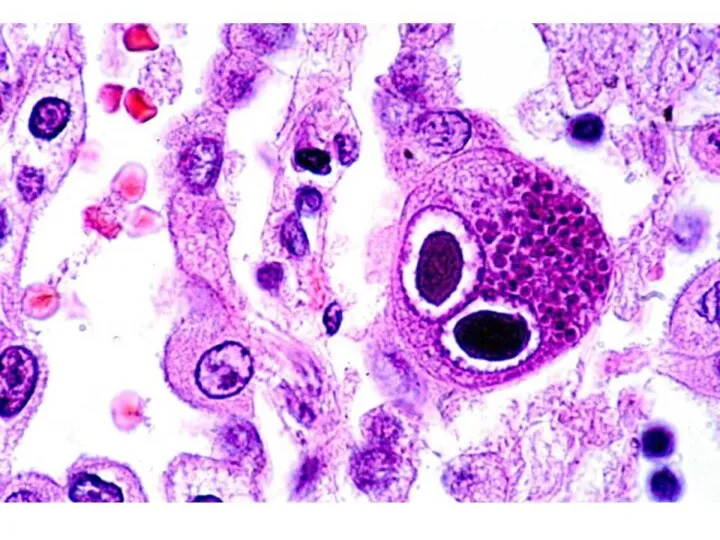
Иммунопатогенез
Репликация вируса происходит в лейкоцитах, клетках системы мононуклеарных фагоцитов.
Процесс репликации заканчивается

Слайд 47Клинические формы
Скрытая форма
Субклиническая форма
Манифестная форма
Со временем – нарастание ИД состояния
Клинические формы
Скрытая форма
Субклиническая форма
Манифестная форма
Со временем – нарастание ИД состояния

Слайд 48Вирус Эпштейна-Барра
Вирус Эпштейна-Барр (Epstein-Barr virus - EBV), так же называемый человеческим герпесвирусом
Вирус Эпштейна-Барра
Вирус Эпштейна-Барр (Epstein-Barr virus - EBV), так же называемый человеческим герпесвирусом

Слайд 49Вирус Эпштейна—Барра
Вирус содержит ДНК; вирион состоит из капсида диаметром 120—150 нм, окруженного
Вирус Эпштейна—Барра
Вирус содержит ДНК; вирион состоит из капсида диаметром 120—150 нм, окруженного

Слайд 50Патогенез
Источник инфекции — больной человек, в том числе и больные стертыми формами
Патогенез
Источник инфекции — больной человек, в том числе и больные стертыми формами

 Чума XXI века многоликий герпес
Чума XXI века многоликий герпес Иммунопрофилактика. Календарь прививок
Иммунопрофилактика. Календарь прививок Бронхиальная астма у детей
Бронхиальная астма у детей Интерпретация компьютерной томографии
Интерпретация компьютерной томографии Питание лиц пожилого и старческого возраста
Питание лиц пожилого и старческого возраста Влияние образа жизни на индивидуальное и общественное здоровье. Семья и формирование ЗОЖ
Влияние образа жизни на индивидуальное и общественное здоровье. Семья и формирование ЗОЖ Близнецы. Сиамские близнецы
Близнецы. Сиамские близнецы Дистоция презентация
Дистоция презентация Стабилизаторы настроения
Стабилизаторы настроения Практикум по ЭКГ
Практикум по ЭКГ Гельминтозы. Санитарно-гигиенические мероприятия
Гельминтозы. Санитарно-гигиенические мероприятия Эффективность коррекционной развивающей программы на основе игры в боча для лиц с дцп
Эффективность коррекционной развивающей программы на основе игры в боча для лиц с дцп Система гемостаза у беременных и акушерские патологии
Система гемостаза у беременных и акушерские патологии Травмы, язвы
Травмы, язвы Мастер-класс Стрижка ногтей пациенту
Мастер-класс Стрижка ногтей пациенту Сексология. Нормальная сексология
Сексология. Нормальная сексология Проба на индивидуальную совместимость по системе АВО
Проба на индивидуальную совместимость по системе АВО Сестринский уход при заболеваниях и травмах прямой кишки
Сестринский уход при заболеваниях и травмах прямой кишки Обморожение
Обморожение Оптико-акустический метод регистрации динамики растворения наночастиц кремния при лазерном облучении
Оптико-акустический метод регистрации динамики растворения наночастиц кремния при лазерном облучении Статистика COVID-19 в зависимости от процента вакцинированных
Статистика COVID-19 в зависимости от процента вакцинированных “Медицинская деятельность ровесница первого человека”, - писал И. П. Павлов
“Медицинская деятельность ровесница первого человека”, - писал И. П. Павлов Средства коррекции иммунных реакций
Средства коррекции иммунных реакций TLAKOVÉ BODY A PUNKCE CÉV HLAVY A KRKU, JEJICH PALPACE
TLAKOVÉ BODY A PUNKCE CÉV HLAVY A KRKU, JEJICH PALPACE Профилактика инфекционных заболеваний: ангины
Профилактика инфекционных заболеваний: ангины Нарушение кровотока, и обусловленная этим недостаточность газообмена в легких
Нарушение кровотока, и обусловленная этим недостаточность газообмена в легких Шизофрения. Симптомы шизофрении
Шизофрения. Симптомы шизофрении Лечебная физическая культура
Лечебная физическая культура