- Половой хроматин и клинико-патофизиологическое значение его определения. Тема 2

Содержание
- 2. Цель занятия: выявление полового хроматина *в ядрах соматических клеток человека: - слизистой буккального эпителия; - нейтрофилов
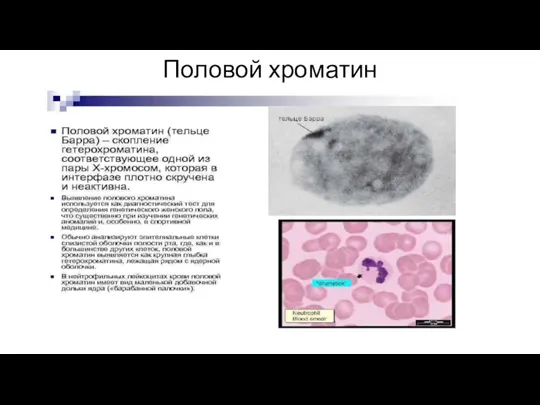
- 3. Половой хроматин (тельце Барра) — материал генетически инактивированной X-хромосомы, в норме выявляемый только в соматических (диплоидных)
- 4. В клетках различных тканей (в различных популяциях) половой хроматин выявляется с неодинаковой частотой: 80—90 % нервных
- 5. Половой хроматин локализуется в ядре, чаще всего прилегая к ядерной оболочке, с которой он связан тонкими
- 6. Все организмы, в кариотипе которых имеются две Х-хромосомы, содержат одно тельце полового хроматина. При трисомии по
- 7. Объекты исследования Х-хроматина — клетки буккального эпителия, эпителиальные клетки мочевого осадка, нейтрофилы крови и др. В
- 8. Изучение полового хроматина используется для верификации хромосомного пола (1), диагностики хромосомных аберраций по половым хромосомам (2),
- 9. На 5—6-й день эмбрионального развития сначала в клетках трофоэктодермы, затем — в других частях зародыша осуществляется
- 10. Механизм этого явления связан с метилированием ДНК инактивируемой хромосомы. Специальный участок Xq27.3 на Х-хромосоме («островок CpG»)
- 11. У человека развитие мужского пола предопределено наличием Y-хромосомы. В присутствии любого количества Х-хромосом одной Y-хромосомы достаточно
- 12. Ген SRY, находящийся в Y-хромосоме, срабатывает на 6—7-й неделе эмбриогенеза, запуская в действие ряд генов, локализованных
- 13. Продукция тестостерона и антимюллерова ингибирующего пептида в гонадах плода предопределяет (2—3-й месяц эмбриогенеза) формирование внутренних половых
- 14. Метаболит андрогенов (5-альфа-дигидротестостерон), не конвертируемый в эстрогены, контролирует маскулинный тип формирования наружных гениталий на 3—4-м месяце
- 15. При наличии Y-хромосомы и высокой продукции тестостерона обеспечивается высокое содержание метаболитов эстрогенов, подавляющих формирование обратной связи
- 16. Это ведет к установлению мужского нециклического типа гипоталамо-гипофизарной регуляции половых функций и обеспечивает на 4—6-м месяце
- 17. Созревание центров секреции гонадотропинов идет под контролем эстрогенов, полученных из тестостерона, а центров, определяющих половое влечение,
- 18. При отсутствии гена SRY, даже если кариотип 46ХХ, а не 45X0 (синдром Шерешевского — Тернера), описанные
- 19. Под наследственными понимают заболевания с первичными техническими дефектами в программном аппарате клеток, передаваемые по наследству через
- 20. Моногенные наследственные заболевания контролируются одним геном и поэтому наследуются в соответствии с законами Менделя (например, гемофилия
- 21. Полигенные наследственные болезни наследуются по аддитиво-полигенному типу, как правило, с пороговым эффектом по воздействию того или
- 22. Важно помнить, что мутации, хотя и лежат в основе патогенеза наследственных болезней, но не тождественны самому
- 23. Работа 1. Определение полового хроматина в соскобе со слизистой полости рта
- 24. Перед взятием соскоба со слизистой щеки несколько раз зубами сжать слизистую, затем проглотить слюну и шпателем
- 25. Не давая материалу подсохнуть, наносят на мазок 1—2 капли 1 % уксуснокислого орсеина. При отсутствии орсеина
- 26. Через 1—2 мин на мазок накладывается покровное стекло, а поверх него — кусочек фильтровальной бумаги, сложенной
- 27. Половой хроматин
- 28. Слева: клетки буккального эпителия. Красная стрелка указывает на глыбку полового хроматина (тельце Барра). Справа: Ядро фибробласта
- 29. Работа 2. Нахождение Х-хроматина в нейтрофилах крови человека Окрашенный мазок крови просматривают под иммерсионным увеличением, внимательно
- 30. Половой хроматин
- 32. Ход анализа полового хроматина: 1. Получение клеточного материала. Источник – разнообразные ткани, но предпочтительны те, что
- 33. 2. Фиксация препаратов раствором метанола или смесью этанола и уксусной кислоты (3:1) или исключительно этанолом. 3.
- 34. 5. Окрашивание полового хроматина. Методы окраски X- и Y-хроматина различны. Х - хроматин окрашивается препаратами на
- 35. Работа 3. Скрининг-тесты наследственных обменных болезней человека А. Определение фенилаланина в моче человека (проба И. А.
- 36. Фенилкетонурия (ФКУ) – это нарушение метаболизма аминокислот, приводящее к возникновению клинического синдрома умственной отсталости с когнитивными
- 38. фенилаланин – незаменимая аминокислота, строительный блок белков. При фенилкетонурии (наследстевнная ферментопатия, дефект гена РАН (12q23.2; ген
- 39. При своевременной диагностике можно полностью избежать патологических изменений, если с рождения и до полового созревания ограничить
- 40. При рождении ребёнка в роддомах на 3—4 сутки берут анализ крови и проводят неонатальный скрининг для
- 41. В. Определение гомогентизиновой кислоты в моче человека (проба А. Гаррода) К 0,5 мл мочи больного алкаптонурией
- 42. Алкапто́нури́я — наследственное заболевание, обусловленное выпадением функций оксидазы гомогентизиновой кислоты и характеризующееся расстройством обмена тирозина и
- 43. Концепция метаболического блока de facto основана на принципе «один ген - один фермент - один признак
- 44. Согласно концепции метаболического блока, при наследственном заболевании имеется дефицит белка-фермента. Это ведет к нарушению определенной биохимической
- 45. Избыток субстрата может вовлекаться в альтернативные превращения, давая такие продукты, которые отсутствуют или имеются лишь в
- 46. Помимо избытка субстрата и действия альтернативных продуктов ( которые могут быть токсическими), часть симптомов наследственных болезней
- 47. Современная трактовка принципа Бидла-Тейтама и положений Гарода о метаболическом блоке сильно изменилась по сравнению с оригинальной,
- 48. 2. Не все белки, кодируемые генами - ферменты. Множество наследственных болезней не связано с дефектом какого
- 49. 3. Один белок кодируется чаще всего не одним геном, а несколькими, причем каждый ответственен за структуру
- 50. Алкаптонурия возникает вследствие мутации гена, кодирующего синтез оксидазы гомогентезиновой кислоты. Данная патология характеризуется аутосомно-рецессивным типом наследования.
- 51. В нормальных условиях гомогентизиновая кислота — промежуточный продукт распада тирозина и фенилаланина — переводится в малеилацетоуксусную
- 52. Из-за дефекта фермента этот процесс тормозится, и остающаяся в избытке гомогентизиновая кислота превращается полифенолоксидазой в хиноновый
- 53. Алкаптон - если он не полностью экскретируется с мочой - откладывается в хрящевой и другой соединительной
- 54. Г. Определение меди в моче человека Пробу мочи больного гепатоцеребральной дистрофией (болезнь Вильсона — Коновалова) нанносят
- 55. Болезнь Вильсона — Коновалова (Гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона) — наследственное нарушение метаболизма
- 56. Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание передаётся аутосомно-рецессивно. Ген ATP7B,
- 57. Аутосомно-рецессивный тип наследования болезни Вильсона. 25 % вероятность рождения больного у родителей-гетерозигот
- 58. Английский невролог Сэмюель Вильсон ( S. Wilson, (1878 - 1937) в 1912 году описал у больных:
- 59. Было предположено, что ген PRNP (кодирует прионный белок, активен в головном мозге и других тканях) участвует
- 60. Медь в организме выполняет множество функций. В основном она является кофактором для таких ферментов, как церулоплазмин,
- 61. Часть меди связывается с металлотионеином, а другая — перемещается в аппарат Гольджи с помощью транспортного белка
- 62. В гепатоцитах белок ATP7B 1) связывает медь с церулоплазмином и высвобождает его в кровь, 2) удаляет
- 63. Когда меди в печени становится больше, чем белков её связывающих, происходит их окислительное повреждение . Это
- 64. Основную роль в патогенезе играет накопление меди в нервной ткани (особенно поражены базальные ганглии), почках, печени
- 65. Церулоплазмин участвует в процессе выведения меди из организма. В печени формируется крупноузловой или смешанный цирроз. В
- 66. Кольца Кайзера-Флейшера: желтовато-зелёная или зеленовато-коричневая пигментация по периферии роговицы на десцеметовой мембране (задняя пограничная мембрана). Кольца
- 67. Лечение Патогенетическое лечение направлено на выведение меди из организма. Для этого применяются комплексообразующие соединения: тиолы, пеницилламин.
- 70. Скачать презентацию
Слайд 2Цель занятия:
выявление полового хроматина
*в ядрах соматических клеток человека:
- слизистой буккального
Цель занятия: выявление полового хроматина *в ядрах соматических клеток человека: - слизистой буккального

Слайд 3Половой хроматин (тельце Барра) — материал генетически инактивированной X-хромосомы,
в норме выявляемый
Половой хроматин (тельце Барра) — материал генетически инактивированной X-хромосомы, в норме выявляемый

Слайд 4В клетках различных тканей (в различных популяциях) половой хроматин выявляется с неодинаковой
В клетках различных тканей (в различных популяциях) половой хроматин выявляется с неодинаковой

Слайд 5Половой хроматин локализуется в ядре, чаще всего прилегая к ядерной оболочке, с
Половой хроматин локализуется в ядре, чаще всего прилегая к ядерной оболочке, с

Слайд 6Все организмы, в кариотипе которых
имеются две Х-хромосомы,
содержат одно тельце полового
Все организмы, в кариотипе которых имеются две Х-хромосомы, содержат одно тельце полового

Слайд 7Объекты исследования Х-хроматина — клетки буккального эпителия, эпителиальные клетки мочевого осадка, нейтрофилы
Объекты исследования Х-хроматина — клетки буккального эпителия, эпителиальные клетки мочевого осадка, нейтрофилы

Слайд 8Изучение полового хроматина используется для верификации хромосомного пола (1),
диагностики хромосомных аберраций
Изучение полового хроматина используется для верификации хромосомного пола (1), диагностики хромосомных аберраций

Слайд 9На 5—6-й день эмбрионального развития сначала в клетках трофоэктодермы, затем — в
На 5—6-й день эмбрионального развития сначала в клетках трофоэктодермы, затем — в

Слайд 10Механизм этого явления связан с метилированием ДНК инактивируемой хромосомы.
Специальный участок Xq27.3
Механизм этого явления связан с метилированием ДНК инактивируемой хромосомы. Специальный участок Xq27.3

Слайд 11
У человека развитие мужского пола предопределено наличием Y-хромосомы.
В присутствии любого количества
У человека развитие мужского пола предопределено наличием Y-хромосомы. В присутствии любого количества

Слайд 12Ген SRY, находящийся в Y-хромосоме, срабатывает на 6—7-й неделе эмбриогенеза,
запуская в
Ген SRY, находящийся в Y-хромосоме, срабатывает на 6—7-й неделе эмбриогенеза, запуская в

Слайд 13
Продукция тестостерона и антимюллерова ингибирующего пептида в гонадах плода
предопределяет
(2—3-й месяц
Продукция тестостерона и антимюллерова ингибирующего пептида в гонадах плода предопределяет (2—3-й месяц

Слайд 14 Метаболит андрогенов
(5-альфа-дигидротестостерон),
не конвертируемый в эстрогены,
контролирует маскулинный тип формирования
Метаболит андрогенов (5-альфа-дигидротестостерон), не конвертируемый в эстрогены, контролирует маскулинный тип формирования

Слайд 15При наличии Y-хромосомы и высокой продукции тестостерона
обеспечивается высокое содержание метаболитов эстрогенов,
При наличии Y-хромосомы и высокой продукции тестостерона обеспечивается высокое содержание метаболитов эстрогенов,

Слайд 16Это ведет к установлению мужского нециклического типа гипоталамо-гипофизарной регуляции половых функций
и
Это ведет к установлению мужского нециклического типа гипоталамо-гипофизарной регуляции половых функций и

Слайд 17Созревание центров секреции гонадотропинов идет под контролем эстрогенов, полученных из тестостерона,
а
Созревание центров секреции гонадотропинов идет под контролем эстрогенов, полученных из тестостерона, а

Слайд 18При отсутствии гена SRY, даже если кариотип 46ХХ, а не 45X0 (синдром
При отсутствии гена SRY, даже если кариотип 46ХХ, а не 45X0 (синдром

Слайд 19Под наследственными понимают заболевания с
первичными техническими дефектами в программном аппарате клеток,
Под наследственными понимают заболевания с первичными техническими дефектами в программном аппарате клеток,

Слайд 20Моногенные наследственные заболевания контролируются одним геном и поэтому наследуются в соответствии с
Моногенные наследственные заболевания контролируются одним геном и поэтому наследуются в соответствии с

Слайд 21Полигенные наследственные болезни наследуются по
аддитиво-полигенному типу, как правило, с пороговым эффектом
Полигенные наследственные болезни наследуются по аддитиво-полигенному типу, как правило, с пороговым эффектом

Слайд 22Важно помнить, что мутации, хотя и лежат в основе патогенеза наследственных болезней,
Важно помнить, что мутации, хотя и лежат в основе патогенеза наследственных болезней,

Слайд 23Работа 1.
Определение полового хроматина в соскобе
со слизистой полости рта
Работа 1.
Определение полового хроматина в соскобе
со слизистой полости рта

Слайд 24Перед взятием соскоба со слизистой щеки несколько раз зубами сжать слизистую, затем
Перед взятием соскоба со слизистой щеки несколько раз зубами сжать слизистую, затем

Слайд 25Не давая материалу подсохнуть, наносят на мазок 1—2 капли 1 % уксуснокислого
Не давая материалу подсохнуть, наносят на мазок 1—2 капли 1 % уксуснокислого

Слайд 26Через 1—2 мин на мазок накладывается покровное стекло, а поверх него —
Через 1—2 мин на мазок накладывается покровное стекло, а поверх него —

Слайд 27Половой хроматин
Половой хроматин
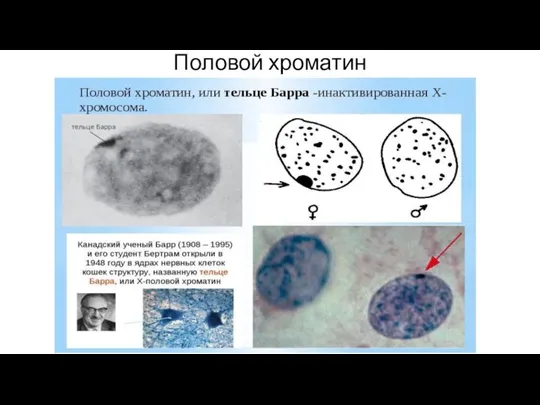
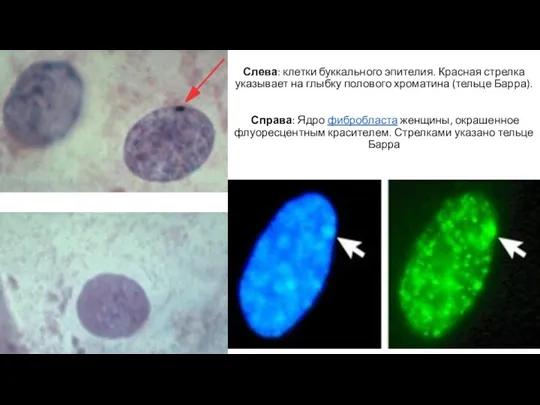
Слайд 28Слева: клетки буккального эпителия. Красная стрелка указывает на глыбку полового хроматина (тельце
Слева: клетки буккального эпителия. Красная стрелка указывает на глыбку полового хроматина (тельце
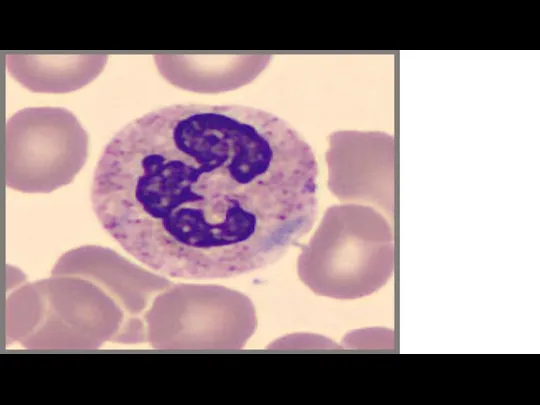
Слайд 29Работа 2. Нахождение Х-хроматина в нейтрофилах крови человека
Окрашенный мазок крови просматривают под
Работа 2. Нахождение Х-хроматина в нейтрофилах крови человека Окрашенный мазок крови просматривают под

Слайд 30Половой хроматин
Половой хроматин
Слайд 32
Ход анализа полового хроматина:
1. Получение клеточного материала.
Источник – разнообразные ткани, но
Ход анализа полового хроматина: 1. Получение клеточного материала. Источник – разнообразные ткани, но

Слайд 332. Фиксация препаратов раствором метанола или смесью этанола и уксусной кислоты (3:1)
2. Фиксация препаратов раствором метанола или смесью этанола и уксусной кислоты (3:1)

Слайд 345. Окрашивание полового хроматина.
Методы окраски X- и Y-хроматина различны. Х -
5. Окрашивание полового хроматина. Методы окраски X- и Y-хроматина различны. Х -

Слайд 35Работа 3. Скрининг-тесты наследственных обменных болезней человека
А. Определение фенилаланина в моче человека
Работа 3. Скрининг-тесты наследственных обменных болезней человека А. Определение фенилаланина в моче человека

Слайд 36Фенилкетонурия (ФКУ) – это нарушение метаболизма аминокислот, приводящее к возникновению клинического синдрома умственной
Фенилкетонурия (ФКУ) – это нарушение метаболизма аминокислот, приводящее к возникновению клинического синдрома умственной


Слайд 38 фенилаланин – незаменимая аминокислота, строительный блок белков.
При фенилкетонурии (наследстевнная ферментопатия, дефект
фенилаланин – незаменимая аминокислота, строительный блок белков. При фенилкетонурии (наследстевнная ферментопатия, дефект

Слайд 39При своевременной диагностике можно полностью избежать патологических изменений, если с рождения и
При своевременной диагностике можно полностью избежать патологических изменений, если с рождения и

Слайд 40При рождении ребёнка в роддомах
на 3—4 сутки берут анализ крови и проводят неонатальный скрининг для
При рождении ребёнка в роддомах на 3—4 сутки берут анализ крови и проводят неонатальный скрининг для

Слайд 41В. Определение гомогентизиновой кислоты в моче человека (проба А. Гаррода)
К 0,5 мл
В. Определение гомогентизиновой кислоты в моче человека (проба А. Гаррода) К 0,5 мл

Слайд 42Алкапто́нури́я — наследственное заболевание, обусловленное выпадением функций оксидазы гомогентизиновой кислоты и характеризующееся расстройством обмена тирозина и экскрецией с
Алкапто́нури́я — наследственное заболевание, обусловленное выпадением функций оксидазы гомогентизиновой кислоты и характеризующееся расстройством обмена тирозина и экскрецией с

Слайд 43Концепция метаболического блока de facto основана на принципе
«один ген - один
Концепция метаболического блока de facto основана на принципе «один ген - один

Слайд 44Согласно концепции метаболического блока, при наследственном заболевании имеется дефицит белка-фермента.
Это ведет
Согласно концепции метаболического блока, при наследственном заболевании имеется дефицит белка-фермента. Это ведет

Слайд 45
Избыток субстрата может вовлекаться в альтернативные превращения, давая такие продукты, которые отсутствуют
Избыток субстрата может вовлекаться в альтернативные превращения, давая такие продукты, которые отсутствуют

Слайд 46Помимо избытка субстрата и действия альтернативных продуктов
( которые могут быть токсическими),
Помимо избытка субстрата и действия альтернативных продуктов ( которые могут быть токсическими),

Слайд 47Современная трактовка принципа Бидла-Тейтама и положений Гарода о метаболическом блоке сильно изменилась
Современная трактовка принципа Бидла-Тейтама и положений Гарода о метаболическом блоке сильно изменилась

Слайд 482. Не все белки, кодируемые генами - ферменты.
Множество наследственных болезней не
2. Не все белки, кодируемые генами - ферменты. Множество наследственных болезней не

Слайд 493. Один белок кодируется чаще всего не одним геном, а несколькими, причем
3. Один белок кодируется чаще всего не одним геном, а несколькими, причем

Слайд 50Алкаптонурия возникает вследствие мутации гена, кодирующего синтез оксидазы гомогентезиновой кислоты.
Данная патология характеризуется
аутосомно-рецессивным
типом
Алкаптонурия возникает вследствие мутации гена, кодирующего синтез оксидазы гомогентезиновой кислоты. Данная патология характеризуется аутосомно-рецессивным типом

Слайд 51В нормальных условиях гомогентизиновая кислота — промежуточный продукт распада тирозина и фенилаланина
В нормальных условиях гомогентизиновая кислота — промежуточный продукт распада тирозина и фенилаланина

Слайд 52Из-за дефекта фермента этот процесс тормозится, и остающаяся в избытке гомогентизиновая кислота
Из-за дефекта фермента этот процесс тормозится, и остающаяся в избытке гомогентизиновая кислота

Слайд 53Алкаптон - если он не полностью экскретируется с мочой - откладывается в
Алкаптон - если он не полностью экскретируется с мочой - откладывается в

Слайд 54Г. Определение меди в моче человека
Пробу мочи больного гепатоцеребральной дистрофией (болезнь Вильсона
Г. Определение меди в моче человека Пробу мочи больного гепатоцеребральной дистрофией (болезнь Вильсона

Слайд 55Болезнь Вильсона — Коновалова (Гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона) —
наследственное
Болезнь Вильсона — Коновалова (Гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона) — наследственное

Слайд 56Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание
Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание

Слайд 57Аутосомно-рецессивный тип наследования болезни Вильсона.
25 % вероятность рождения больного у родителей-гетерозигот
Аутосомно-рецессивный тип наследования болезни Вильсона.
25 % вероятность рождения больного у родителей-гетерозигот

Слайд 58Английский невролог Сэмюель Вильсон
( S. Wilson, (1878 - 1937) в 1912 году
описал у больных:
* типичные изменения
Английский невролог Сэмюель Вильсон ( S. Wilson, (1878 - 1937) в 1912 году описал у больных: * типичные изменения

Слайд 59Было предположено, что ген PRNP
(кодирует прионный белок, активен в головном мозге
Было предположено, что ген PRNP (кодирует прионный белок, активен в головном мозге

Слайд 60Медь в организме выполняет множество функций.
В основном она является кофактором для
Медь в организме выполняет множество функций. В основном она является кофактором для

Слайд 61Часть меди связывается с металлотионеином, а другая — перемещается в аппарат Гольджи с помощью транспортного белка
Часть меди связывается с металлотионеином, а другая — перемещается в аппарат Гольджи с помощью транспортного белка

Слайд 62
В гепатоцитах белок ATP7B
1) связывает медь с церулоплазмином и высвобождает его в
В гепатоцитах белок ATP7B 1) связывает медь с церулоплазмином и высвобождает его в

Слайд 63Когда меди в печени становится больше,
чем белков её связывающих,
происходит их
Когда меди в печени становится больше, чем белков её связывающих, происходит их

Слайд 64Основную роль в патогенезе играет накопление меди в нервной ткани
(особенно поражены базальные
Основную роль в патогенезе играет накопление меди в нервной ткани (особенно поражены базальные

Слайд 65Церулоплазмин участвует в процессе выведения меди из организма.
В печени формируется крупноузловой
Церулоплазмин участвует в процессе выведения меди из организма. В печени формируется крупноузловой

Слайд 66Кольца Кайзера-Флейшера:
желтовато-зелёная или зеленовато-коричневая пигментация по периферии роговицы на десцеметовой мембране (задняя пограничная мембрана).
Кольца - отложения меди на
Кольца Кайзера-Флейшера: желтовато-зелёная или зеленовато-коричневая пигментация по периферии роговицы на десцеметовой мембране (задняя пограничная мембрана). Кольца - отложения меди на

Слайд 67Лечение
Патогенетическое лечение направлено на выведение меди из организма. Для этого применяются комплексообразующие соединения: тиолы, пеницилламин.
Лечение Патогенетическое лечение направлено на выведение меди из организма. Для этого применяются комплексообразующие соединения: тиолы, пеницилламин.


 Природно – очаговые заболевания на территории центрального черноземья
Природно – очаговые заболевания на территории центрального черноземья Диабетическая ретинопатия
Диабетическая ретинопатия Применение радиационных технологий в медицине
Применение радиационных технологий в медицине Профилактика наркомании
Профилактика наркомании Breast Massager
Breast Massager Сахарный диабет
Сахарный диабет Гены MHC I класса
Гены MHC I класса Шок, признаки шока. Противошоковые мероприятия
Шок, признаки шока. Противошоковые мероприятия Энцефалиты. Классификация энцефалитов
Энцефалиты. Классификация энцефалитов Процесс движения крови по организму человека
Процесс движения крови по организму человека Глаза и зрение. Дневник ощущений
Глаза и зрение. Дневник ощущений Генетика человека с основами медицинской генетики
Генетика человека с основами медицинской генетики Сахарный диабет 1 типа
Сахарный диабет 1 типа Травматический шок
Травматический шок Микробиологическая диагностика дифтерии
Микробиологическая диагностика дифтерии Современные методы гемостаза при кровотечениях из варикозно расширенных вен пищевода и желудка
Современные методы гемостаза при кровотечениях из варикозно расширенных вен пищевода и желудка МДК 02.03 Лекция №5 акушерки ЛМханическая травма
МДК 02.03 Лекция №5 акушерки ЛМханическая травма COVID-19 - Энциклопедия поведения в период пандемии
COVID-19 - Энциклопедия поведения в период пандемии Lektsia_1_2
Lektsia_1_2 Артериальное давление
Артериальное давление Гипопаратиреоз. Клинический разбор
Гипопаратиреоз. Клинический разбор Жизненный цикл ВИЧ
Жизненный цикл ВИЧ Professionalnaya_gigiena
Professionalnaya_gigiena Terminologia anatomica
Terminologia anatomica Анксиолитики (транквилизаторы)
Анксиолитики (транквилизаторы) Гипо және гиперкортицизм
Гипо және гиперкортицизм Гельминтозы. Пути заражения и меры профилактики
Гельминтозы. Пути заражения и меры профилактики Психотропные вещества как производное оружие
Психотропные вещества как производное оружие