- Синдромы микроструктурных аномалий хромосом

Содержание
- 2. Никто из нас не совершенен. Всё больше генетических тестов становится доступно, и каждый из нас, в
- 3. CNV Copy Number Variations Фрагмент ДНК, размером более 1 т.п.н., по числу копий отличающийся от референсного
- 6. Локус-специфичная частота CNV варьирует от 0,0001 до 0,00001. Патогенные дупликации встречаются реже, чем делеции, т.к. их
- 7. Патогенетические механизмы развития заболеваний: изменение дозы генов эффект положения разрыв кодирующей последовательности гена образование химерного гена
- 8. Клиническое значение Точки разрыва аберрации могут нарушать кодирующую последовательность гена или приводить к возникновению нового гибридного
- 9. Динамика описания микроделеционных и микродупликационных синдромов Weiss A. et al. Microdeletion and Microduplication Syndromes J Histochem
- 10. Микроделеция - это утрата участка хромосомы, размеры которого находятся за гранью разрешающей возможности световой микроскопии.
- 11. Микродупликация - наличие дополнительной копии крошечного фрагмента хромосомы.
- 13. Nevado et al. New microdeletion and microduplication syndromes: A comprehensive review Genet Mol Biol. 2014. V.
- 14. СИНДРОМЫ МИКРОДЕЛЕЦИЙ СИНДРОМ ВИЛЬЯМСА (7q11.23) Синдром «лица эльфов» - заболевание, вызванное делецией размером 1,5 – 1,8
- 15. СИНДРОМ ВИЛЬЯМСА (7q11.23) Клинические признаки: Необычное лицо, низкий рост, короткий нос, полные щеки, маленькая нижняя челюсть,
- 16. СИНДРОМ МИКРОДЕЛЕЦИИ СИНДРОМ РУБИНШТЕЙНА-ТЕЙБИ (16p13.3) Заболевание, вызванное делецией или мутациями в генах CREBBP (локус16р13.3 ) или
- 17. СИНДРОМ МИКРОДЕЛЕЦИИ СИНДРОМ РУБИНШТЕЙНА-ТЕЙБИ (16p13.3) Клинические признаки: Микроцефалия, брахицефалия, приподнятые брови,антимонголоидный разрез глазных щелей, постнатальное отставание
- 18. СИНДРОМ МИКРОДЕЛЕЦИИ СИНДРОМ СМИТ-МАГЕНИС (17р11.2) Заболевание, вызванное делецией 3,7 миллионов нуклеотидных пар или мутациями в гене
- 19. СИНДРОМ СМИТ МАГЕНИС Клинические признаки: Черпно-лицевые аномалии: брахицефалия, выпуклый лоб гипоплазия средней части лица, брахидактилия, пороки
- 20. СИНДРОМ МИКРОДУПЛИКАЦИИ 22q11.2 Синдром Ди Джорджи, велокардиофациальный синдром, делеция центрального участка длинного плеча хромосомы в 1,5
- 21. СИНДРОМ МИКРОДЕЛЕЦИИ 22q11.2 Клинические признаки: Имеются пороки сердца, гипоплазия тимуса, гипоплазия паращитовидных желез . Тип наследования
- 22. СИНДРОМЫ ХРОМОСОМНОЙ НЕСТАБИЛЬНОСТИ Синдромы хромосомный нестабильности, также известные как синдромы хромосомной поломки - это группа генетических
- 23. Анемия Фанкони 13 генов, мутации в которых вызывают развитие анемии Фанкони: FANCA, FANCB, FANCC, FANCD1, FANCD2,
- 24. Анемия Фанкони Клинические признаки: Недостаточность костного мозга, небольшой рост, различные повреждения кожи, рук, головы, глаз, почек,
- 25. Ниймеген синдром (8q21) Ген NBN, мутации в котором приводят к развитию синдрома, состоит из 50 тыс.п.н.
- 26. Ниймеген синдром Клинические признаки: Микроцефалия, задержка умственного развития, отсталость физ. Развития, комбинированный иммунодефицит, «птичье лицо» .
- 27. Синдром Блума (15q26.1) Причиной заболевания служит мутация в гене BLM (RECQL3), который имеет 22 экзона. Кодирует
- 28. Синдром Блума Клинические признаки: Не высокий рост, характерные высыпания на коже, высокий голос, специфические черты лица,
- 29. Атаксия телеангиэктазия (синдром Луи-Бара) 11q22-23 Белковый продукт гена ATM относится к семейству фосфатидилинозитолкиназ, контролирует клеточный цикл,
- 30. Атаксия телеангиэктазия Клинические признаки: Мозжечковая атаксия, интенционный тремор, нарушения движений глазного яблока, косоглазие, нистагм, отставание в
- 31. Пигментная ксеродерма Пигментная ксеродерма – генетически разнородное, панэтническое, аутосомно-рецессивное заболевание репарации ДНК. Вызвана мутациями, влияющими на
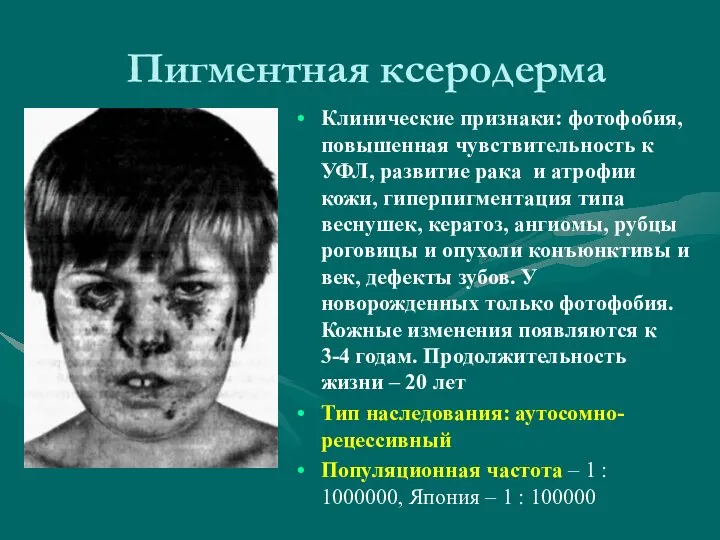
- 32. Пигментная ксеродерма Клинические признаки: фотофобия, повышенная чувствительность к УФЛ, развитие рака и атрофии кожи, гиперпигментация типа
- 34. Скачать презентацию

Слайд 3CNV
Copy Number Variations
Фрагмент ДНК, размером более 1 т.п.н., по числу копий отличающийся
CNV
Copy Number Variations
Фрагмент ДНК, размером более 1 т.п.н., по числу копий отличающийся


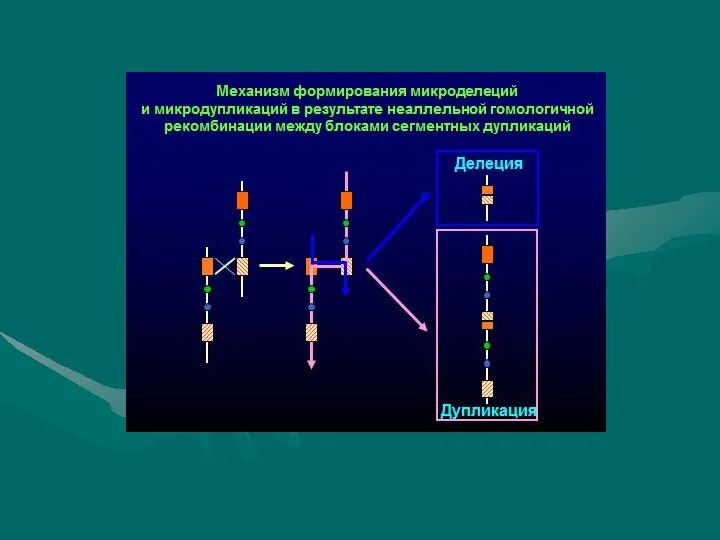
Слайд 6Локус-специфичная частота CNV варьирует от 0,0001 до 0,00001.
Патогенные дупликации встречаются реже, чем
Локус-специфичная частота CNV варьирует от 0,0001 до 0,00001.
Патогенные дупликации встречаются реже, чем

Слайд 7Патогенетические механизмы развития заболеваний:
изменение дозы генов
эффект положения
разрыв кодирующей последовательности гена
образование химерного
Патогенетические механизмы развития заболеваний:
изменение дозы генов
эффект положения
разрыв кодирующей последовательности гена
образование химерного

Слайд 8Клиническое значение
Точки разрыва аберрации могут нарушать кодирующую последовательность гена или приводить к
Клиническое значение
Точки разрыва аберрации могут нарушать кодирующую последовательность гена или приводить к

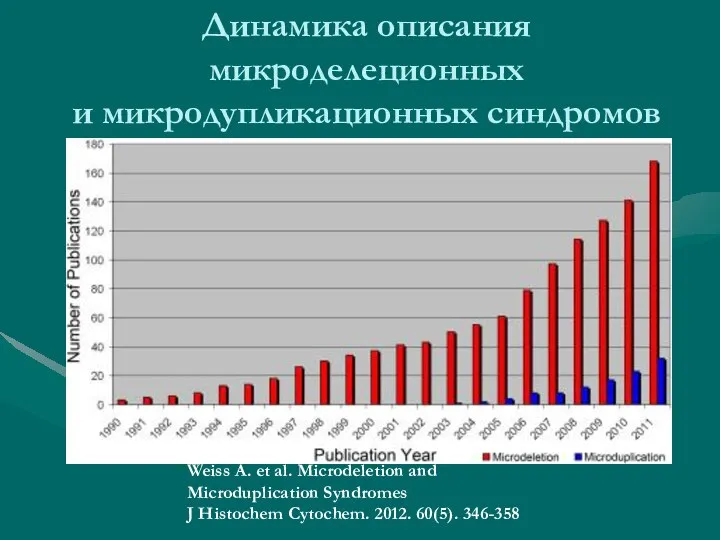
Слайд 9Динамика описания микроделеционных
и микродупликационных синдромов
Weiss A. et al. Microdeletion and Microduplication Syndromes
J
Динамика описания микроделеционных
и микродупликационных синдромов
Weiss A. et al. Microdeletion and Microduplication Syndromes
J
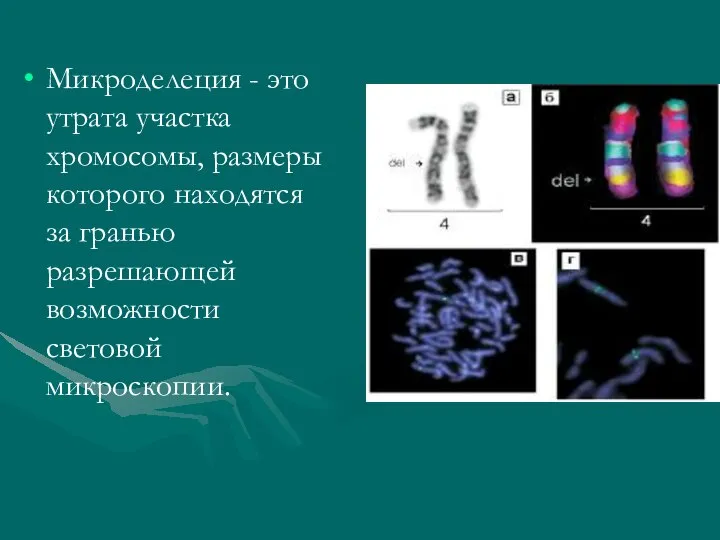
Слайд 10Микроделеция - это утрата участка хромосомы, размеры которого находятся за гранью разрешающей
Микроделеция - это утрата участка хромосомы, размеры которого находятся за гранью разрешающей
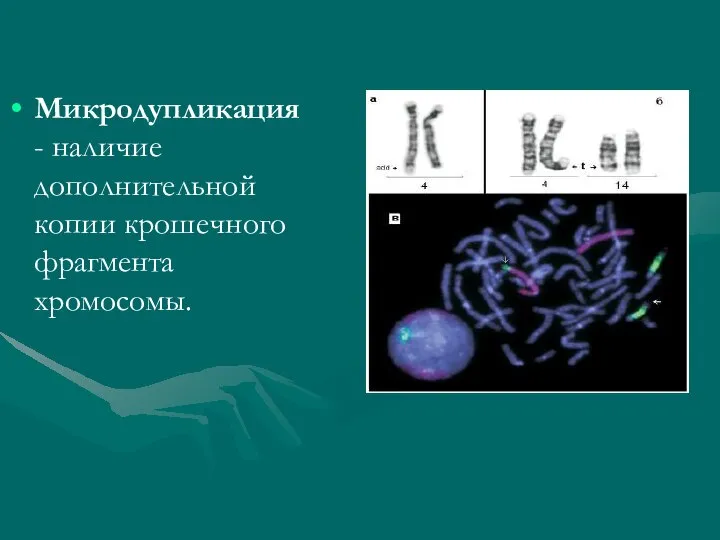
Слайд 11Микродупликация - наличие дополнительной копии крошечного фрагмента хромосомы.
Микродупликация - наличие дополнительной копии крошечного фрагмента хромосомы.
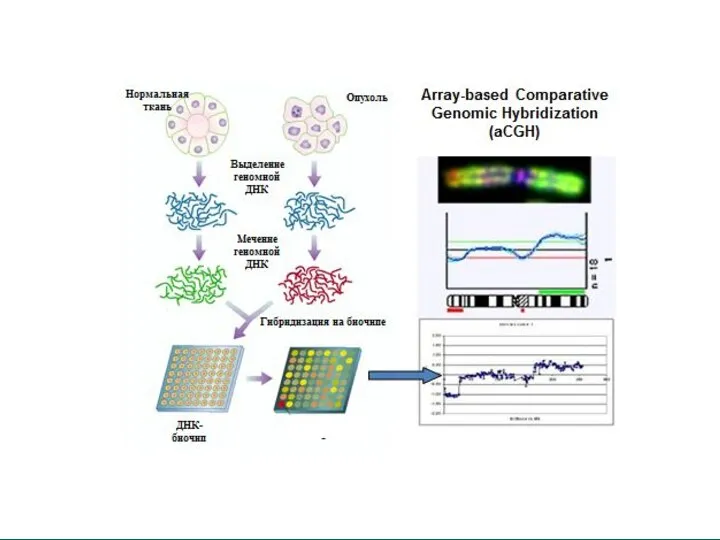
Слайд 13Nevado et al. New microdeletion and microduplication syndromes: A comprehensive review
Genet
Nevado et al. New microdeletion and microduplication syndromes: A comprehensive review
Genet

Слайд 14СИНДРОМЫ МИКРОДЕЛЕЦИЙ
СИНДРОМ ВИЛЬЯМСА (7q11.23)
Синдром «лица эльфов» - заболевание, вызванное делецией размером 1,5
СИНДРОМЫ МИКРОДЕЛЕЦИЙ
СИНДРОМ ВИЛЬЯМСА (7q11.23)
Синдром «лица эльфов» - заболевание, вызванное делецией размером 1,5

Слайд 15СИНДРОМ ВИЛЬЯМСА (7q11.23)
Клинические признаки:
Необычное лицо, низкий рост, короткий нос, полные щеки, маленькая
СИНДРОМ ВИЛЬЯМСА (7q11.23)
Клинические признаки:
Необычное лицо, низкий рост, короткий нос, полные щеки, маленькая

Слайд 16СИНДРОМ МИКРОДЕЛЕЦИИ
СИНДРОМ РУБИНШТЕЙНА-ТЕЙБИ (16p13.3)
Заболевание, вызванное делецией или мутациями в генах CREBBP (локус16р13.3
СИНДРОМ МИКРОДЕЛЕЦИИ
СИНДРОМ РУБИНШТЕЙНА-ТЕЙБИ (16p13.3)
Заболевание, вызванное делецией или мутациями в генах CREBBP (локус16р13.3

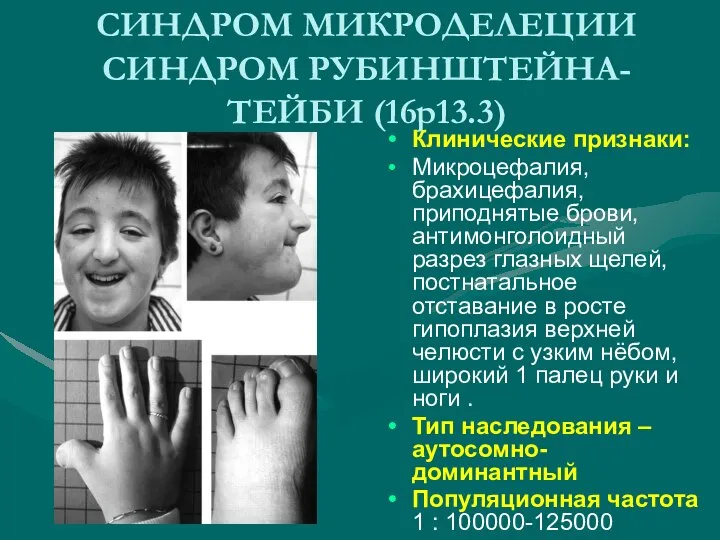
Слайд 17СИНДРОМ МИКРОДЕЛЕЦИИ
СИНДРОМ РУБИНШТЕЙНА-ТЕЙБИ (16p13.3)
Клинические признаки:
Микроцефалия, брахицефалия, приподнятые брови,антимонголоидный разрез глазных щелей, постнатальное
СИНДРОМ МИКРОДЕЛЕЦИИ
СИНДРОМ РУБИНШТЕЙНА-ТЕЙБИ (16p13.3)
Клинические признаки:
Микроцефалия, брахицефалия, приподнятые брови,антимонголоидный разрез глазных щелей, постнатальное
Слайд 18СИНДРОМ МИКРОДЕЛЕЦИИ
СИНДРОМ СМИТ-МАГЕНИС (17р11.2)
Заболевание, вызванное делецией 3,7 миллионов нуклеотидных пар или
СИНДРОМ МИКРОДЕЛЕЦИИ
СИНДРОМ СМИТ-МАГЕНИС (17р11.2)
Заболевание, вызванное делецией 3,7 миллионов нуклеотидных пар или

Слайд 19СИНДРОМ СМИТ МАГЕНИС
Клинические признаки:
Черпно-лицевые аномалии: брахицефалия, выпуклый лоб гипоплазия средней части лица,
СИНДРОМ СМИТ МАГЕНИС
Клинические признаки:
Черпно-лицевые аномалии: брахицефалия, выпуклый лоб гипоплазия средней части лица,

Слайд 20СИНДРОМ МИКРОДУПЛИКАЦИИ
22q11.2
Синдром Ди Джорджи, велокардиофациальный синдром, делеция центрального участка длинного плеча хромосомы
СИНДРОМ МИКРОДУПЛИКАЦИИ
22q11.2
Синдром Ди Джорджи, велокардиофациальный синдром, делеция центрального участка длинного плеча хромосомы

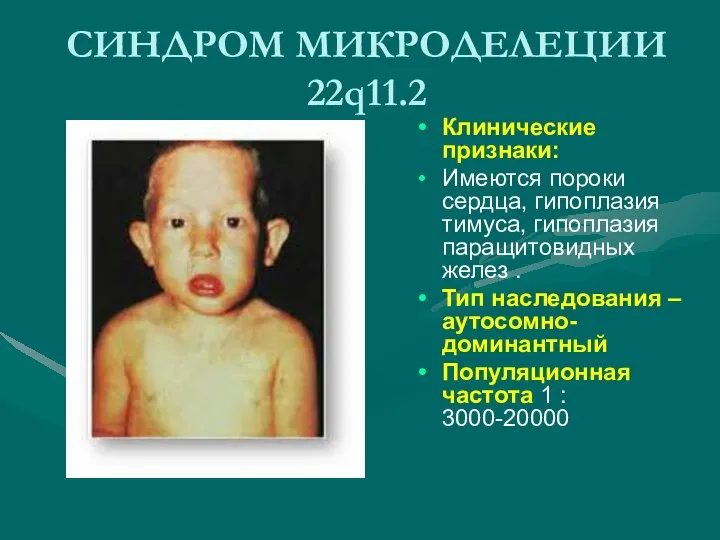
Слайд 21СИНДРОМ МИКРОДЕЛЕЦИИ
22q11.2
Клинические признаки:
Имеются пороки сердца, гипоплазия тимуса, гипоплазия паращитовидных желез .
Тип наследования
СИНДРОМ МИКРОДЕЛЕЦИИ
22q11.2
Клинические признаки:
Имеются пороки сердца, гипоплазия тимуса, гипоплазия паращитовидных желез .
Тип наследования
Слайд 22СИНДРОМЫ ХРОМОСОМНОЙ НЕСТАБИЛЬНОСТИ
Синдромы хромосомный нестабильности, также известные как синдромы хромосомной поломки -
СИНДРОМЫ ХРОМОСОМНОЙ НЕСТАБИЛЬНОСТИ
Синдромы хромосомный нестабильности, также известные как синдромы хромосомной поломки -

Слайд 23Анемия Фанкони
13 генов, мутации в которых вызывают развитие анемии Фанкони: FANCA, FANCB, FANCC, FANCD1,
Анемия Фанкони
13 генов, мутации в которых вызывают развитие анемии Фанкони: FANCA, FANCB, FANCC, FANCD1,

Слайд 24Анемия Фанкони
Клинические признаки:
Недостаточность костного мозга, небольшой рост, различные повреждения кожи, рук, головы,
Анемия Фанкони
Клинические признаки:
Недостаточность костного мозга, небольшой рост, различные повреждения кожи, рук, головы,

Слайд 25Ниймеген синдром (8q21)
Ген NBN, мутации в котором приводят к развитию синдрома, состоит
Ниймеген синдром (8q21)
Ген NBN, мутации в котором приводят к развитию синдрома, состоит

Слайд 26Ниймеген синдром
Клинические признаки:
Микроцефалия, задержка умственного развития, отсталость физ. Развития, комбинированный иммунодефицит, «птичье
Ниймеген синдром
Клинические признаки:
Микроцефалия, задержка умственного развития, отсталость физ. Развития, комбинированный иммунодефицит, «птичье

Слайд 27Синдром Блума (15q26.1)
Причиной заболевания служит мутация в гене BLM (RECQL3), который имеет
Синдром Блума (15q26.1)
Причиной заболевания служит мутация в гене BLM (RECQL3), который имеет

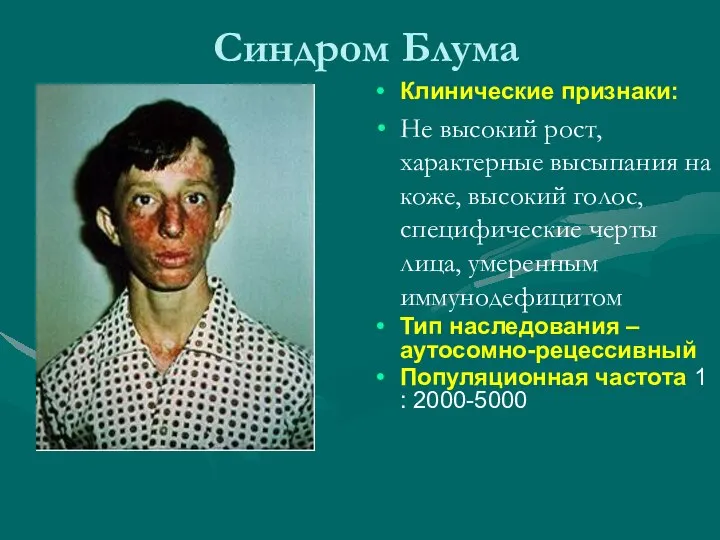
Слайд 28Синдром Блума
Клинические признаки:
Не высокий рост, характерные высыпания на коже, высокий голос, специфические черты
Синдром Блума
Клинические признаки:
Не высокий рост, характерные высыпания на коже, высокий голос, специфические черты
Слайд 29Атаксия телеангиэктазия (синдром Луи-Бара) 11q22-23
Белковый продукт гена ATM относится к семейству фосфатидилинозитолкиназ,
Атаксия телеангиэктазия (синдром Луи-Бара) 11q22-23
Белковый продукт гена ATM относится к семейству фосфатидилинозитолкиназ,

Слайд 30Атаксия телеангиэктазия
Клинические признаки:
Мозжечковая атаксия, интенционный тремор, нарушения движений глазного яблока, косоглазие, нистагм,
Атаксия телеангиэктазия
Клинические признаки:
Мозжечковая атаксия, интенционный тремор, нарушения движений глазного яблока, косоглазие, нистагм,

Слайд 31Пигментная ксеродерма
Пигментная ксеродерма – генетически разнородное, панэтническое, аутосомно-рецессивное заболевание репарации ДНК.
Вызвана мутациями,
Пигментная ксеродерма
Пигментная ксеродерма – генетически разнородное, панэтническое, аутосомно-рецессивное заболевание репарации ДНК.
Вызвана мутациями,

Слайд 32Пигментная ксеродерма
Клинические признаки: фотофобия, повышенная чувствительность к УФЛ, развитие рака и атрофии
Пигментная ксеродерма
Клинические признаки: фотофобия, повышенная чувствительность к УФЛ, развитие рака и атрофии
 Геморрагические диатезы лекция 22
Геморрагические диатезы лекция 22 Антибиотики. Ципрофлоксацины, амоксициллины
Антибиотики. Ципрофлоксацины, амоксициллины Анализ ведения больных с КЭ 2013-2015гг по материалам ГБУЗ НСО ГИКБ №1
Анализ ведения больных с КЭ 2013-2015гг по материалам ГБУЗ НСО ГИКБ №1 аэробные инфекции
аэробные инфекции Антифосфолипидті синдром кезіндегі диагностикалық критерилері және емі
Антифосфолипидті синдром кезіндегі диагностикалық критерилері және емі Общая этиология и общий патогенез эндокринных нарушений
Общая этиология и общий патогенез эндокринных нарушений Нарушения внешнего дыхания
Нарушения внешнего дыхания Инсульт в молодом возрасте
Инсульт в молодом возрасте Задание в тестовой форме на тему Аритмии
Задание в тестовой форме на тему Аритмии Чума. История
Чума. История Болезни почек. Виды повреждений
Болезни почек. Виды повреждений Наборы дезинфекции антисептики. Санитайзеры. Увлажнители. Защита. Позитив
Наборы дезинфекции антисептики. Санитайзеры. Увлажнители. Защита. Позитив Гриппозное поражение нервной системы
Гриппозное поражение нервной системы Основы микробиологии, санитарии и гигиены
Основы микробиологии, санитарии и гигиены Жұқпалы аурулар
Жұқпалы аурулар Моноклонды антиденелер, алу тәсілдері, иммундыдиагностика және иммундытерапиядағы ролі
Моноклонды антиденелер, алу тәсілдері, иммундыдиагностика және иммундытерапиядағы ролі Колликвативный туберкулёз кожи (скрофулодерма)
Колликвативный туберкулёз кожи (скрофулодерма) Химиотерапевтические препараты. Антибиотики. Противовирусные средства
Химиотерапевтические препараты. Антибиотики. Противовирусные средства Влияние мобильных устройств на организм человека
Влияние мобильных устройств на организм человека Отчет по производственной практике. Специальность: 34.02.01 Сестринское дело
Отчет по производственной практике. Специальность: 34.02.01 Сестринское дело Гемофилия қандай ауруға жатады
Гемофилия қандай ауруға жатады Коронавирус. Законодательные и локальные нормативные акты
Коронавирус. Законодательные и локальные нормативные акты Детская инфекционная болезнь корь
Детская инфекционная болезнь корь тератология1 см
тератология1 см Учение об инфекции. Биологический метод исследования
Учение об инфекции. Биологический метод исследования Порядок проведения диспансеризации определенных групп взрослого населения
Порядок проведения диспансеризации определенных групп взрослого населения Лечение рака щитовидной железы
Лечение рака щитовидной железы Известные люди с заиканием
Известные люди с заиканием