- Наследственность и патология

Содержание
- 2. Содержание: Наследственные болезни Врожденные пороки развития Хромосомные болезни Моногенные болезни Диагностика наследственных болезней
- 3. Наследственные болезни Термин «наследственная болезнь» подразумевает унаследованное от родителей (полученное с одной из родительских гамет или
- 4. Классификация наследственных заболеваний В настоящее время чаще всего используется классификация наследственных болезней, предложенная Н.П. Бочковым (1984).
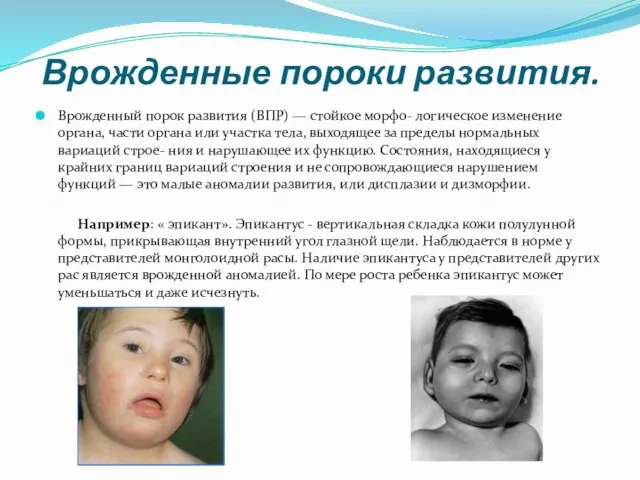
- 5. Врожденные пороки развития. Врожденный порок развития (ВПР) — стойкое морфо- логическое изменение органа, части органа или
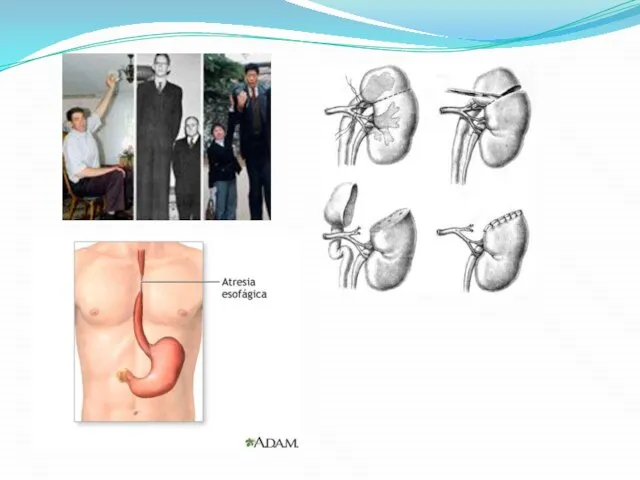
- 6. Виды наиболее распространенных ВПР Макросомия (гигантизм) — увеличение длины,массы тела. Атрезия — отсутствие естественного канала, отверстия.
- 8. Хромосомные болезни Хромосомные болезни, или синдромы, — это группа врожденных патологических состояний, вызываемых изменениями числа (геномные
- 9. Классификация хромосомных болезней Классификация хромосомных болезней основывается на следующих критериях: — типе мутации (полная или частичная
- 10. Синдром Дауна К наиболее распространенным заболеваниям с количественным нарушением хромосом относится трисомия 21 (наличие 47 хромосом
- 11. У детей с синдромом дауна отмечаются черепно-лицевые аномалии: уплощение затылка, эпикант, монголоидный разрез глаз, короткий нос,
- 12. Синдром Вильямса Для синдрома Вильямса (синдром «лицо эльфа») характерны полные, опущенные вниз щеки, нередко голубоватые склеры,
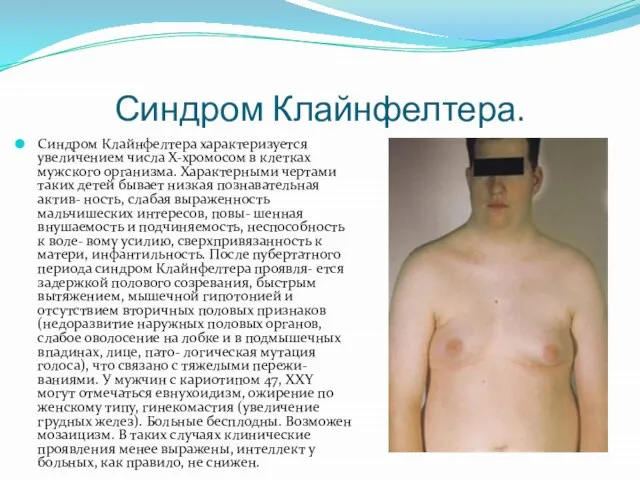
- 13. Синдром Клайнфелтера. Синдром Клайнфелтера характеризуется увеличением числа Х-хромосом в клетках мужского организма. Характерными чертами таких детей
- 14. Синдром 47, XYY Может отмечаться фенотипическое сходство с синдромом Клайнфелтера: высокий рост, евнуховидность. Неглубокая умственная отсталость
- 15. Моногенные болезни Моногенные болезни — это разнородная по клиническим проявлениям, этиологии и патогенезу группа заболеваний, наследующихся
- 16. Классификация моногенных болезней Классификация моногенных болезней Исходя из генетического принципа, выделяют болезни с аутосомно–доминантным, аутосомно-рецессивным, Х-сцепленным
- 17. Синдром Ваарденбурга Дети с синдромом Ваарденбурга характеризуются особенностями внешности, связанными с нарушениями пигментации: седая прядь или
- 18. Синдром Сотоса Характерными его признаками являются макроцефалия, выступающие лобные бугры, увеличенные кисти и стопы, гипертелоризм, антимонголоидный
- 19. Синдром Дубовица характеризуется врожденной и постнатальной гипотрофией, соматической ослабленностью, отставанием психического развития, нарушениями пищеварения. Одним из
- 20. Синдром Барде-Бидля Характерно выраженное ожирение с обильным отложением жира на лице, груди, животе, ягодицах и бедрах.
- 21. Диагностика наследственных болезней Диагностика отклонений в развитии является важным этапом консультирования и не направлена на социальную
- 22. Методы диагностики наследственных форм отклонений в развитии Многие наследственные формы патологии диагностируются по особенностям внешнего облика,
- 23. Чаще всего привлекает внимание своеобразное строение лица: Кукольное лицо с длинными ресницами характерно для детей с
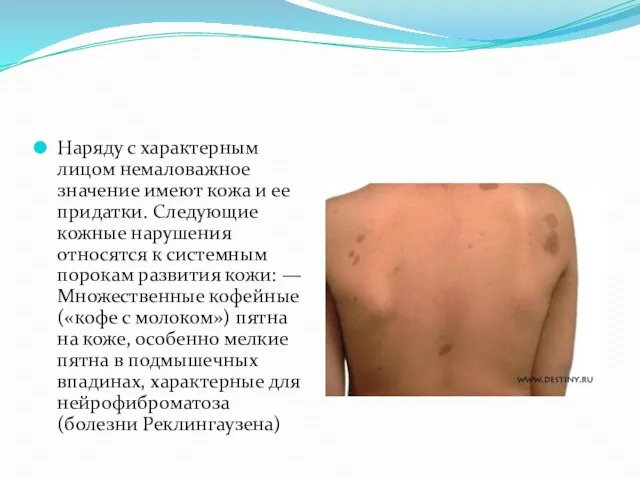
- 24. Наряду с характерным лицом немаловажное значение имеют кожа и ее придатки. Следующие кожные нарушения относятся к
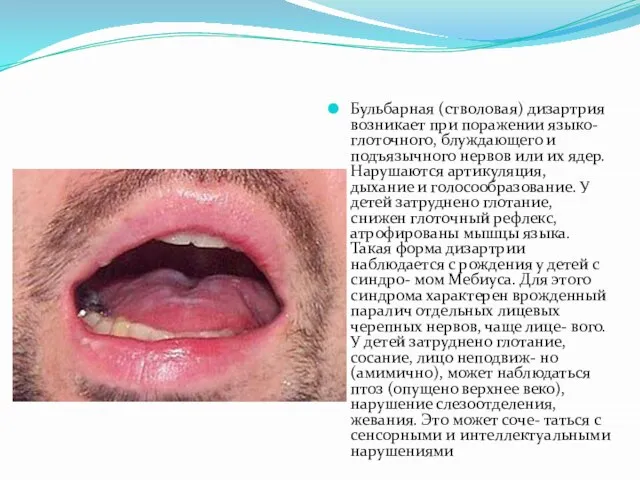
- 25. Бульбарная (стволовая) дизартрия возникает при поражении языко-глоточного, блуждающего и подъязычного нервов или их ядер. Нарушаются артикуляция,
- 27. Скачать презентацию
Слайд 2Содержание:
Наследственные болезни
Врожденные пороки развития
Хромосомные болезни
Моногенные болезни
Диагностика наследственных болезней
Содержание:
Наследственные болезни
Врожденные пороки развития
Хромосомные болезни
Моногенные болезни
Диагностика наследственных болезней

Слайд 3Наследственные болезни
Термин «наследственная болезнь» подразумевает унаследованное от родителей (полученное с одной из
Наследственные болезни
Термин «наследственная болезнь» подразумевает унаследованное от родителей (полученное с одной из

Слайд 4Классификация наследственных заболеваний
В настоящее время чаще всего используется классификация наследственных болезней, предложенная
Классификация наследственных заболеваний
В настоящее время чаще всего используется классификация наследственных болезней, предложенная

Слайд 5Врожденные пороки развития.
Врожденный порок развития (ВПР) — стойкое морфо- логическое изменение органа, части
Врожденные пороки развития.
Врожденный порок развития (ВПР) — стойкое морфо- логическое изменение органа, части
Слайд 6Виды наиболее распространенных ВПР
Макросомия (гигантизм) — увеличение длины,массы тела.
Атрезия — отсутствие естественного
Виды наиболее распространенных ВПР
Макросомия (гигантизм) — увеличение длины,массы тела.
Атрезия — отсутствие естественного

Слайд 8Хромосомные болезни
Хромосомные болезни, или синдромы, — это группа врожденных патологических состояний, вызываемых
Хромосомные болезни
Хромосомные болезни, или синдромы, — это группа врожденных патологических состояний, вызываемых

Слайд 9Классификация хромосомных болезней
Классификация хромосомных болезней основывается на следующих критериях:
— типе мутации (полная
Классификация хромосомных болезней
Классификация хромосомных болезней основывается на следующих критериях:
— типе мутации (полная

Слайд 10Синдром Дауна
К наиболее распространенным заболеваниям с количественным нарушением хромосом относится трисомия 21
Синдром Дауна
К наиболее распространенным заболеваниям с количественным нарушением хромосом относится трисомия 21

Слайд 11У детей с синдромом дауна отмечаются черепно-лицевые аномалии: уплощение затылка, эпикант, монголоидный
У детей с синдромом дауна отмечаются черепно-лицевые аномалии: уплощение затылка, эпикант, монголоидный

Слайд 12Синдром Вильямса
Для синдрома Вильямса (синдром «лицо эльфа») характерны полные, опущенные вниз щеки,
Синдром Вильямса
Для синдрома Вильямса (синдром «лицо эльфа») характерны полные, опущенные вниз щеки,

Слайд 13Синдром Клайнфелтера.
Синдром Клайнфелтера характеризуется увеличением числа Х-хромосом в клетках мужского организма. Характерными
Синдром Клайнфелтера.
Синдром Клайнфелтера характеризуется увеличением числа Х-хромосом в клетках мужского организма. Характерными
Слайд 14Синдром 47, XYY
Может отмечаться фенотипическое сходство с синдромом Клайнфелтера: высокий рост, евнуховидность.
Синдром 47, XYY
Может отмечаться фенотипическое сходство с синдромом Клайнфелтера: высокий рост, евнуховидность.

Слайд 15Моногенные болезни
Моногенные болезни — это разнородная по клиническим проявлениям, этиологии и патогенезу
Моногенные болезни
Моногенные болезни — это разнородная по клиническим проявлениям, этиологии и патогенезу

Слайд 16Классификация моногенных болезней
Классификация моногенных болезней Исходя из генетического принципа, выделяют болезни с
Классификация моногенных болезней
Классификация моногенных болезней Исходя из генетического принципа, выделяют болезни с

Слайд 17 Синдром Ваарденбурга
Дети с синдромом Ваарденбурга характеризуются особенностями внешности, связанными с нарушениями пигментации:
Синдром Ваарденбурга
Дети с синдромом Ваарденбурга характеризуются особенностями внешности, связанными с нарушениями пигментации:

Слайд 18 Синдром Сотоса
Характерными его признаками являются макроцефалия, выступающие лобные бугры, увеличенные кисти и
Синдром Сотоса
Характерными его признаками являются макроцефалия, выступающие лобные бугры, увеличенные кисти и

Слайд 19Синдром Дубовица
характеризуется врожденной и постнатальной гипотрофией, соматической ослабленностью, отставанием психического развития, нарушениями
Синдром Дубовица
характеризуется врожденной и постнатальной гипотрофией, соматической ослабленностью, отставанием психического развития, нарушениями

Слайд 20 Синдром Барде-Бидля
Характерно выраженное ожирение с обильным отложением жира на лице, груди,
Синдром Барде-Бидля
Характерно выраженное ожирение с обильным отложением жира на лице, груди,

Слайд 21Диагностика наследственных болезней
Диагностика отклонений в развитии является важным этапом консультирования и не
Диагностика наследственных болезней
Диагностика отклонений в развитии является важным этапом консультирования и не

Слайд 22Методы диагностики наследственных форм отклонений в развитии
Многие наследственные формы патологии диагностируются по
Методы диагностики наследственных форм отклонений в развитии
Многие наследственные формы патологии диагностируются по

Слайд 23Чаще всего привлекает внимание своеобразное строение лица: Кукольное лицо с длинными ресницами
Чаще всего привлекает внимание своеобразное строение лица: Кукольное лицо с длинными ресницами

Слайд 24Наряду с характерным лицом немаловажное значение имеют кожа и ее придатки. Следующие
Наряду с характерным лицом немаловажное значение имеют кожа и ее придатки. Следующие
Слайд 25Бульбарная (стволовая) дизартрия возникает при поражении языко-глоточного, блуждающего и подъязычного нервов или
Бульбарная (стволовая) дизартрия возникает при поражении языко-глоточного, блуждающего и подъязычного нервов или
 История развития психопатологии в зарубежных странах
История развития психопатологии в зарубежных странах Ветсансараптау әдістері
Ветсансараптау әдістері Шизофрения. Продуктивная симптоматика
Шизофрения. Продуктивная симптоматика Entamoeba Histolytica
Entamoeba Histolytica Микобактерия туберкулеза
Микобактерия туберкулеза Избыточная масса тела. Ожирение
Избыточная масса тела. Ожирение Пищеварительная система. Врожденные дефекты лица
Пищеварительная система. Врожденные дефекты лица Данные исследования физиологической нормы крови иммунологических и биохимических показателей при заболеваниях пародонта у детей
Данные исследования физиологической нормы крови иммунологических и биохимических показателей при заболеваниях пародонта у детей Гуморальные факторы неспецифического иммунитета ротовой полости
Гуморальные факторы неспецифического иммунитета ротовой полости Здоровое питание - залог здоровья человека, фундамент его счастья
Здоровое питание - залог здоровья человека, фундамент его счастья Гипоксия. Гипоксические состояния
Гипоксия. Гипоксические состояния Уреаплазмоз. Микоплазмоз
Уреаплазмоз. Микоплазмоз Особенности репродукции человека в связи с его биосоциальной сущностью
Особенности репродукции человека в связи с его биосоциальной сущностью Организация медицинского обслуживания в дошкольных образовательных учреждениях
Организация медицинского обслуживания в дошкольных образовательных учреждениях Первая помощь при ушибах, переломах, растяжениях и вывихах
Первая помощь при ушибах, переломах, растяжениях и вывихах Препарат ШМ-54. Соскоб с шейки матки
Препарат ШМ-54. Соскоб с шейки матки Внутренняя среда организма. Кровь и её состав
Внутренняя среда организма. Кровь и её состав Особенности кровоснабжения и иннервации ЧЛО в детском возрасте
Особенности кровоснабжения и иннервации ЧЛО в детском возрасте Гиперосмолярная кома
Гиперосмолярная кома Раны. Виды ран. Первая медицинская помощь
Раны. Виды ран. Первая медицинская помощь Острые респираторные заболевания и грипп
Острые респираторные заболевания и грипп Заболевания почек
Заболевания почек Пуринергиялық синапстарда қозудың берілуіне әсер ететін дәрілер
Пуринергиялық синапстарда қозудың берілуіне әсер ететін дәрілер Цистоскопия, артроскопия, торакоскопия, лапароскопия, кольпоскопия
Цистоскопия, артроскопия, торакоскопия, лапароскопия, кольпоскопия Фармацевтический рынок Украины Состояние и тенденции развития О.Л. Добранчук Руководитель аналитической службы компании МОРИОН
Фармацевтический рынок Украины Состояние и тенденции развития О.Л. Добранчук Руководитель аналитической службы компании МОРИОН  Костномозговое кроветворение и оценка миелограммы
Костномозговое кроветворение и оценка миелограммы Организация специализированной амбулаторнополиклинической помощи населению
Организация специализированной амбулаторнополиклинической помощи населению Первичный туалет новорожденного
Первичный туалет новорожденного