- Наследственные энзимопатии

Содержание
- 2. План Энзимопатии Причины возникновений наследственных энзимопатий Энзимопатии обмена аминокислот Энзимопатии обмена углеводов Энзимопатии обмена липидов Энзимопатии
- 3. Энзимопатии (ферментопатии) в широком смысле слова — патологические изменения активности ферментов. В более узком смысле этим
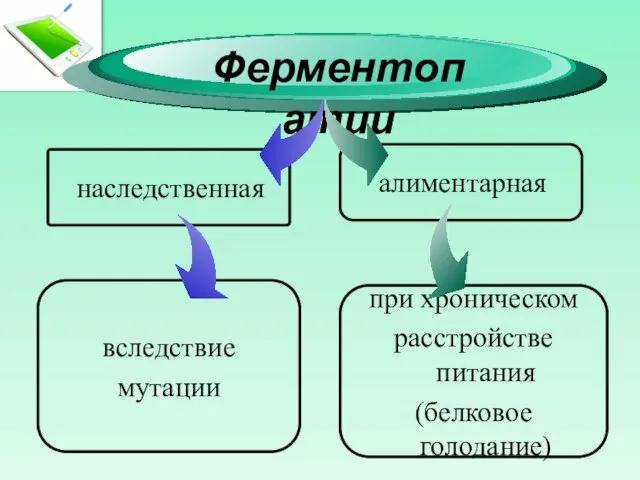
- 4. Ферментопатии наследственная алиментарная при хроническом расстройстве питания (белковое голодание) вследствие мутации
- 5. Наследственные болезни —заболевания человека, обусловленные хромосомными и генными мутациями Наследственные энзимопатии связаны с генетически обусловленной недостаточностью
- 6. Причины возникновения энзимопатий .
- 7. Особенностью течения наследственных энзимопатий является наличие скрытого периода, когда болезнь не имеет выраженных клин. симптомов, но
- 8. Наследственные болезни обмена аминокислот, напр., фенилкетонурия (дефект ферментов, превращающих фенилаланин в тирозин) и гистидинемия (недостаточность фермента,
- 9. Фенилкетонурия Впервые описал A. Foiling в 1934 году. Поражение ЦНС вызывается недостаточностью фермента гидроксилазы-4-фенилаланина, управляющего превращением
- 10. Альбинизм. Блокада активности фермента тирозиназы, катализирующей синтез меланина из тирозина через дигидроксифенилаланин Основными проявлениями ее служат
- 11. Девочка-альбинос из Гондураса
- 12. Лечение 1 Ограничение в диете белка и соответствующей аминокислоты. 2. Дополнительное назначение незаменимых аминокислот. 3. Назначение
- 13. К наследственным болезням углеводного обмена относят гликогенозы, галактоземию, нек-рые формы диабета сахарного и др. Наследственные болезни
- 14. Галактоземия Описана в 1908 году, однако дефект обмена, ее обуславливающий, был открыт лишь в 1956 году.
- 15. Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не усваивается, а промежуточный продукт обмена,
- 16. Проявляется вскоре после рождения у ребенка: отказом от пищи, поносом, рвотой, непереносимостью голода, падением массы тела,
- 17. Наследственные болезни пуринового и пиримидинового обмена включают некоторые формы подагры, синдром Леша-Найхана (возникает при недостаточности гипоксантин
- 18. Наследственными болезнями обмена соединительной ткани являются мукополисахаридозы, Марфана синдром, хондродистрофия; наследственными болезнями крови и кроветворных органов
- 19. Описан G. Gurler в 1919 году. Встречается с частотой — 1: 40 000. Существует еще 15
- 20. Проявляется на первом году жизни. Внешний вид больных — увеличенная голова, выдающиеся лобные бугры, почти отсутствующая
- 21. Основное значение в диагностике наследственных энзимопатий имеют биохимические методы исследования: определение активности ферментов, продуктов обмена веществ,
- 22. Список литературы: 1. Клиническая биохимия: учеб. Пособие для вузов/ под ред. В. А. Ткачука. -2-е изд.,
- 24. Скачать презентацию
Слайд 2План
Энзимопатии
Причины возникновений наследственных энзимопатий
Энзимопатии обмена аминокислот
Энзимопатии обмена углеводов
Энзимопатии обмена липидов
Энзимопатии соединительной ткани
Литература
План
Энзимопатии
Причины возникновений наследственных энзимопатий
Энзимопатии обмена аминокислот
Энзимопатии обмена углеводов
Энзимопатии обмена липидов
Энзимопатии соединительной ткани
Литература

Слайд 3Энзимопатии (ферментопатии) в широком смысле слова — патологические изменения активности ферментов.
В
Энзимопатии (ферментопатии) в широком смысле слова — патологические изменения активности ферментов.
В

Слайд 4Ферментопатии
наследственная
алиментарная
при хроническом
расстройстве питания
(белковое голодание)
вследствие
мутации
Ферментопатии
наследственная
алиментарная
при хроническом
расстройстве питания
(белковое голодание)
вследствие
мутации
Слайд 5Наследственные болезни —заболевания человека, обусловленные хромосомными и генными мутациями
Наследственные энзимопатии связаны с
Наследственные болезни —заболевания человека, обусловленные хромосомными и генными мутациями
Наследственные энзимопатии связаны с

Слайд 6Причины возникновения энзимопатий
.
Причины возникновения энзимопатий
.

Слайд 7Особенностью течения наследственных энзимопатий является наличие скрытого периода, когда болезнь не имеет
Особенностью течения наследственных энзимопатий является наличие скрытого периода, когда болезнь не имеет

Слайд 8Наследственные болезни обмена аминокислот, напр., фенилкетонурия (дефект ферментов, превращающих фенилаланин в тирозин)
Наследственные болезни обмена аминокислот, напр., фенилкетонурия (дефект ферментов, превращающих фенилаланин в тирозин)

Слайд 9Фенилкетонурия
Впервые описал A. Foiling в 1934 году.
Поражение ЦНС вызывается недостаточностью
Фенилкетонурия
Впервые описал A. Foiling в 1934 году.
Поражение ЦНС вызывается недостаточностью

Слайд 10Альбинизм. Блокада активности фермента тирозиназы, катализирующей синтез меланина из тирозина через дигидроксифенилаланин
Основными
Альбинизм. Блокада активности фермента тирозиназы, катализирующей синтез меланина из тирозина через дигидроксифенилаланин
Основными

Слайд 11Девочка-альбинос из Гондураса
Девочка-альбинос из Гондураса

Слайд 12Лечение
1 Ограничение в диете белка и соответствующей аминокислоты.
2. Дополнительное назначение незаменимых аминокислот.
3.
Лечение
1 Ограничение в диете белка и соответствующей аминокислоты. 2. Дополнительное назначение незаменимых аминокислот. 3.

Слайд 13К наследственным болезням углеводного обмена относят гликогенозы, галактоземию, нек-рые формы диабета сахарного
К наследственным болезням углеводного обмена относят гликогенозы, галактоземию, нек-рые формы диабета сахарного

Слайд 14Галактоземия
Описана в 1908 году,
однако дефект обмена,
ее обуславливающий,
был открыт лишь
Галактоземия
Описана в 1908 году,
однако дефект обмена,
ее обуславливающий,
был открыт лишь

Слайд 15 Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не
Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не

Слайд 16Проявляется вскоре после рождения у ребенка:
отказом от пищи, поносом, рвотой, непереносимостью голода,
Проявляется вскоре после рождения у ребенка:
отказом от пищи, поносом, рвотой, непереносимостью голода,

Слайд 17Наследственные болезни пуринового и пиримидинового обмена
включают
некоторые формы
подагры,
Наследственные болезни пуринового и пиримидинового обмена
включают
некоторые формы
подагры,

Слайд 18Наследственными болезнями обмена соединительной ткани являются мукополисахаридозы, Марфана синдром, хондродистрофия; наследственными болезнями
Наследственными болезнями обмена соединительной ткани являются мукополисахаридозы, Марфана синдром, хондродистрофия; наследственными болезнями

Слайд 19Описан G. Gurler в 1919 году.
Встречается с частотой — 1: 40 000.
Описан G. Gurler в 1919 году.
Встречается с частотой — 1: 40 000.

Слайд 20Проявляется на первом году жизни.
Внешний вид больных — увеличенная голова, выдающиеся лобные
Проявляется на первом году жизни.
Внешний вид больных — увеличенная голова, выдающиеся лобные

Слайд 21Основное значение в диагностике наследственных энзимопатий имеют биохимические методы исследования:
определение активности
Основное значение в диагностике наследственных энзимопатий имеют биохимические методы исследования:
определение активности

Слайд 22Список литературы:
1. Клиническая биохимия: учеб. Пособие для вузов/ под ред. В. А.
Список литературы:
1. Клиническая биохимия: учеб. Пособие для вузов/ под ред. В. А.

 Ошибки при изготовлении съёмных протезов
Ошибки при изготовлении съёмных протезов Наследственная недостаточность Альфа-1-антитрипсина
Наследственная недостаточность Альфа-1-антитрипсина Асфиксия новорожденных
Асфиксия новорожденных Возрастные особенности развития сердца у детей. Нарушения. Профилактика
Возрастные особенности развития сердца у детей. Нарушения. Профилактика Отравление метгемоглобинобразователями
Отравление метгемоглобинобразователями Подагра. Диагностика
Подагра. Диагностика Острая ревматическая лихорадка (ОРЛ)
Острая ревматическая лихорадка (ОРЛ) Хирургическая анатомия поджелудочной железы
Хирургическая анатомия поджелудочной железы Роль научно-технического прогресса и урбанизации для патологии современного человека. Новые этиологические факторы болезней
Роль научно-технического прогресса и урбанизации для патологии современного человека. Новые этиологические факторы болезней Микробиологическая диагностика энтеробактерий
Микробиологическая диагностика энтеробактерий Риккетсия диагностикасы
Риккетсия диагностикасы Организация первичной медико- санитарной помощи (ПМСП)
Организация первичной медико- санитарной помощи (ПМСП) Перезагрузка сердца
Перезагрузка сердца Печень. Общий вид печени
Печень. Общий вид печени Медициналық суреттер галереясы
Медициналық суреттер галереясы Egyptian legalacts regarding health care of disabled people & children
Egyptian legalacts regarding health care of disabled people & children Тест Какой у меня обмен веществ?
Тест Какой у меня обмен веществ? Ишемическая болезнь сердца и инфаркт миокарда. (Тема 27)
Ишемическая болезнь сердца и инфаркт миокарда. (Тема 27) Прегравидарная подготовка женщин с синдром поликистозных яичников
Прегравидарная подготовка женщин с синдром поликистозных яичников Цирроз печени
Цирроз печени Вливания. Внутриартериальное вливание
Вливания. Внутриартериальное вливание Особенности визуальной диагностики патологии дыхательной системы у детей на примере пневмонии
Особенности визуальной диагностики патологии дыхательной системы у детей на примере пневмонии Адам баласының денесінде жұдырықтай ет бар
Адам баласының денесінде жұдырықтай ет бар Изменения функций нервной системы при беременности
Изменения функций нервной системы при беременности Жедел холангит
Жедел холангит Помощь при электротравме
Помощь при электротравме Atos BullSequana S and Edge for Retail and Transport
Atos BullSequana S and Edge for Retail and Transport Диагностика хламидиоза. Техника взятия материала
Диагностика хламидиоза. Техника взятия материала