Наследственные синдромы, сопровождающиеся низким ростом (синдром Нунан, Корнелии де Ланге, Вильямса)
- Наследственные синдромы, сопровождающиеся низким ростом (синдром Нунан, Корнелии де Ланге, Вильямса)

Содержание
- 2. Синдром Нунан — мультисистемное аутосомно-доминантное заболевание. Частота в популяции составляет, по разным оценкам, от 1:1000 до
- 3. Клиническая картина заболевания напоминает проявления синдрома Шерешевского-Тёрнера, поэтому раньше это заболевание называлось “Тёрнеровский фенотип с нормальным
- 4. Синдром Нунан является второй по распространенности синдромальной причиной врожденных заболеваний сердца, уступая только трисомии 21 хромосомы.
- 5. Причиной развития данного заболевания являются нарушения в системе универсального клеточного сигнального пути, что объединяет синдром Нунан
- 6. Результаты инструментальных обследований: ЭКГ — ритм синусовый, НРС нет. Признаки увеличения обоих предсердий и перегрузки обоих
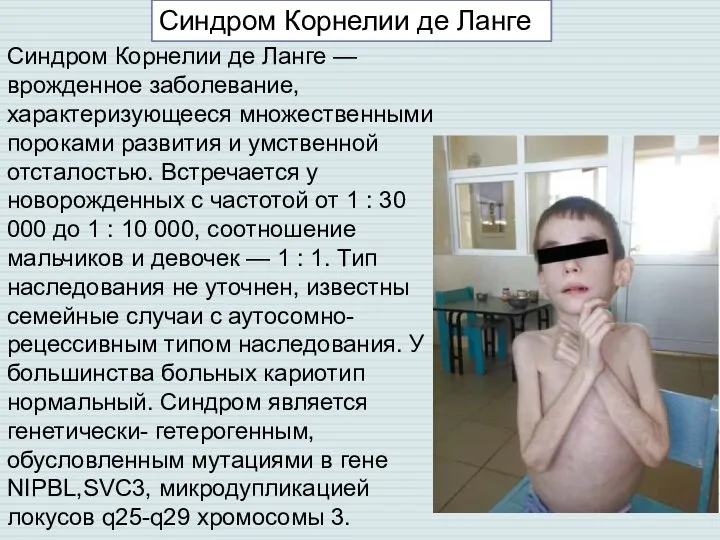
- 7. Синдром Корнелии де Ланге — врожденное заболевание, характеризующееся множественными пороками развития и умственной отсталостью. Встречается у
- 8. В клинической картине синдрома: — аномалии развития черепа, глаз, ушей, лица, зубов, туловища, конечностей, внутренних органов;
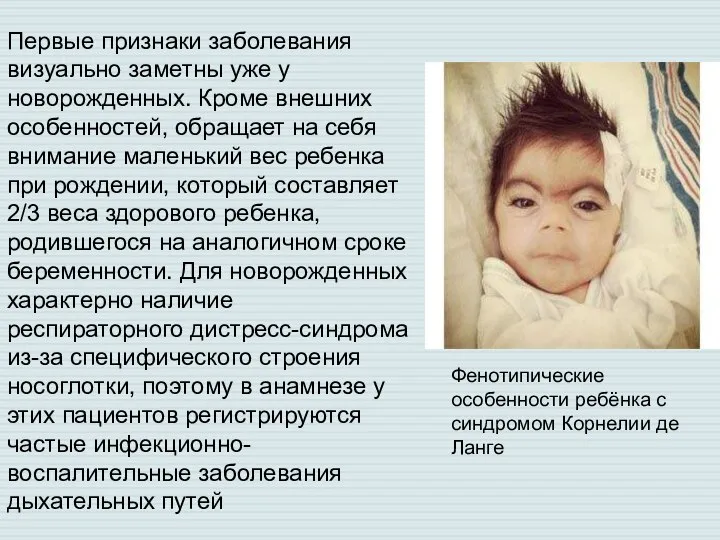
- 9. Первые признаки заболевания визуально заметны уже у новорожденных. Кроме внешних особенностей, обращает на себя внимание маленький
- 10. Диагноз устанавливают на основании фенотипа, исследования кариотипа и методов цитогенетического анализа. Специфического лечения не существует. Применяют
- 11. В последнее время предполагают влияние на развитие данной патологии ряда следующих факторов риска: Наличие в семейном
- 12. Синдром Вильямса (Уильямса) – предположительно аутосомно-доминантное заболевание, вызванное мутацией в гене эластина, картированном на хромосоме 7g
- 13. Дети, страдающие синдромом Вильямса, имеют характерный вид лица, которое внешне напоминает лицо эльфа, отсюда появилось второе
- 14. Различные современные методы исследований позволили установить, что причиной синдрома Вильямса является делеция в 7-й хромосоме. В
- 15. Эластиновая артериопатия присутствует у 75– 80% пациентов с синдромом Вильямса и может поражать любую артерию. Надклапанный
- 16. Специфической терапии не существует. Пациентам проводится симптоматическое лечение и коррекционно-воспитательная работа. Прогноз относительно благоприятный, возможна частичная
- 18. Скачать презентацию
Слайд 2Синдром Нунан — мультисистемное аутосомно-доминантное заболевание. Частота в популяции составляет, по разным
Синдром Нунан — мультисистемное аутосомно-доминантное заболевание. Частота в популяции составляет, по разным

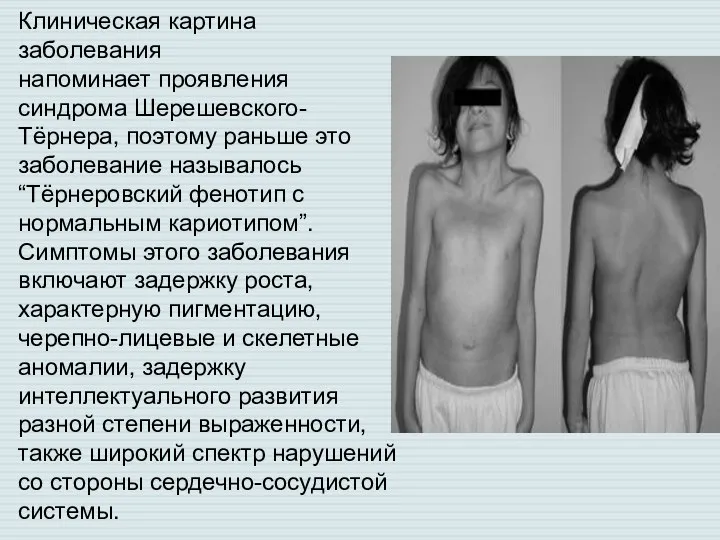
Слайд 3Клиническая картина заболевания
напоминает проявления синдрома Шерешевского-Тёрнера, поэтому раньше это заболевание называлось “Тёрнеровский
Клиническая картина заболевания
напоминает проявления синдрома Шерешевского-Тёрнера, поэтому раньше это заболевание называлось “Тёрнеровский
Слайд 4Синдром Нунан является второй по распространенности синдромальной причиной врожденных заболеваний
сердца, уступая только
Синдром Нунан является второй по распространенности синдромальной причиной врожденных заболеваний
сердца, уступая только

Слайд 5Причиной развития данного заболевания являются нарушения в системе универсального клеточного сигнального пути,
Причиной развития данного заболевания являются нарушения в системе универсального клеточного сигнального пути,

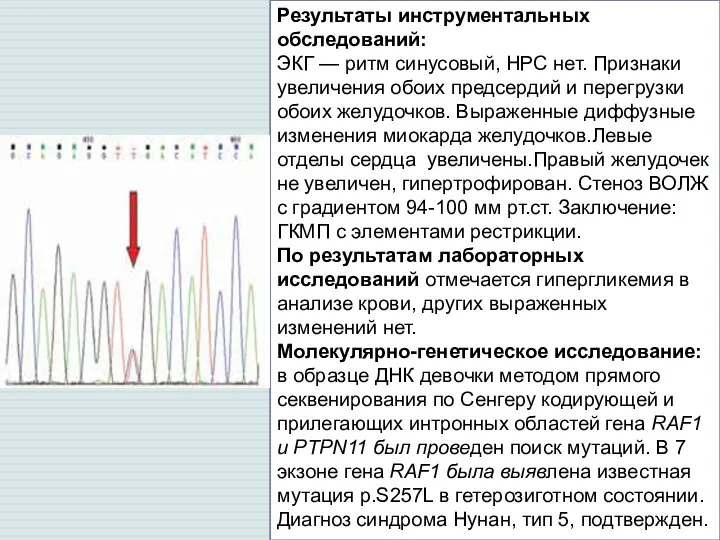
Слайд 6Результаты инструментальных обследований:
ЭКГ — ритм синусовый, НРС нет. Признаки увеличения обоих предсердий
Результаты инструментальных обследований:
ЭКГ — ритм синусовый, НРС нет. Признаки увеличения обоих предсердий
Слайд 7Синдром Корнелии де Ланге — врожденное заболевание, характеризующееся множественными пороками развития и
Синдром Корнелии де Ланге — врожденное заболевание, характеризующееся множественными пороками развития и
Слайд 8В клинической картине синдрома:
— аномалии развития черепа, глаз, ушей, лица, зубов,
туловища, конечностей,
В клинической картине синдрома:
— аномалии развития черепа, глаз, ушей, лица, зубов,
туловища, конечностей,

Слайд 9Первые признаки заболевания визуально заметны уже у новорожденных. Кроме внешних особенностей, обращает
Первые признаки заболевания визуально заметны уже у новорожденных. Кроме внешних особенностей, обращает
Слайд 10Диагноз устанавливают на основании фенотипа, исследования кариотипа и методов цитогенетического
анализа. Специфического лечения
Диагноз устанавливают на основании фенотипа, исследования кариотипа и методов цитогенетического
анализа. Специфического лечения

Слайд 11В последнее время предполагают влияние на развитие данной патологии ряда следующих факторов
В последнее время предполагают влияние на развитие данной патологии ряда следующих факторов

Слайд 12Синдром Вильямса (Уильямса) – предположительно аутосомно-доминантное заболевание, вызванное мутацией в гене эластина,
Синдром Вильямса (Уильямса) – предположительно аутосомно-доминантное заболевание, вызванное мутацией в гене эластина,

Слайд 13Дети, страдающие синдромом Вильямса, имеют характерный вид лица, которое внешне напоминает лицо
Дети, страдающие синдромом Вильямса, имеют характерный вид лица, которое внешне напоминает лицо

Слайд 14Различные современные методы исследований позволили установить, что причиной синдрома Вильямса является делеция

Слайд 15Эластиновая артериопатия присутствует у 75– 80% пациентов с синдромом Вильямса и может
Эластиновая артериопатия присутствует у 75– 80% пациентов с синдромом Вильямса и может

Слайд 16Специфической терапии не существует. Пациентам проводится симптоматическое лечение и коррекционно-воспитательная работа. Прогноз
Специфической терапии не существует. Пациентам проводится симптоматическое лечение и коррекционно-воспитательная работа. Прогноз

 Альбинизм
Альбинизм Fat Embolism Syndrome
Fat Embolism Syndrome Лікарські засоби, що впливають на обмін речовин. Гормональні препарати
Лікарські засоби, що впливають на обмін речовин. Гормональні препарати Использование лекарственного растительного сырья, влияющего на мочевыделительную систему
Использование лекарственного растительного сырья, влияющего на мочевыделительную систему Микроальбуминурия. Результаты диагностики микроальбуминурии по анализу мочи
Микроальбуминурия. Результаты диагностики микроальбуминурии по анализу мочи Признаки зрелости доношенного новорожденнного. Вакцинация
Признаки зрелости доношенного новорожденнного. Вакцинация Анестезия. Общие вопросы и стандартные методики. Практика 4
Анестезия. Общие вопросы и стандартные методики. Практика 4 Физиология иммунной системы. Часть вторая. Единая нейро-иммунно-эндокринная система адаптации организма
Физиология иммунной системы. Часть вторая. Единая нейро-иммунно-эндокринная система адаптации организма Gavvia Brain Enhancer – Works To Make A Fast And Sharp Memory
Gavvia Brain Enhancer – Works To Make A Fast And Sharp Memory ДВС-синдром у беременных. Геморагический шок. Эмболия околоплодными водами
ДВС-синдром у беременных. Геморагический шок. Эмболия околоплодными водами Addiction to drugs is becoming more and more common every day. Drugs among the young people is a problem which affects the whole society and future generations. - презентация_
Addiction to drugs is becoming more and more common every day. Drugs among the young people is a problem which affects the whole society and future generations. - презентация_ Черепные нервы. Орган обоняния. Орган вкуса
Черепные нервы. Орган обоняния. Орган вкуса Научная и практическая деятельность в рамках кружка СНО
Научная и практическая деятельность в рамках кружка СНО Гипертонические препараты
Гипертонические препараты Priekšlikumi alkoholisko dzērienu patēriņa mazināšanai un alkoholisma ierobežošanai 2012. – 2014
Priekšlikumi alkoholisko dzērienu patēriņa mazināšanai un alkoholisma ierobežošanai 2012. – 2014 Антиоксидантный статус крови и ткани глиом с различной степенью злокачественности
Антиоксидантный статус крови и ткани глиом с различной степенью злокачественности Диффузды токсиқалық зоб
Диффузды токсиқалық зоб Хронический бронхит
Хронический бронхит Сестринский уход при травмах
Сестринский уход при травмах Всероссийская олимпиада по детской хирургии. Викторина
Всероссийская олимпиада по детской хирургии. Викторина Sakharny_diabet
Sakharny_diabet Клуб Здоровье
Клуб Здоровье Применение ЛС в аллергологии у беременных и в период лактации
Применение ЛС в аллергологии у беременных и в период лактации Врождённые аномалии развития носа и околоносовых пазух
Врождённые аномалии развития носа и околоносовых пазух Северо-Западный государственный медицинский университет им. И.И. Мечникова
Северо-Западный государственный медицинский университет им. И.И. Мечникова Первая помощь при ранениях, переломах костей, ожогах, отморожениях (тема 2)
Первая помощь при ранениях, переломах костей, ожогах, отморожениях (тема 2) Выявление информативных параметров сигналов электронной аускультации для определения характера шумов дыхания
Выявление информативных параметров сигналов электронной аускультации для определения характера шумов дыхания Профилактика коронавируса
Профилактика коронавируса