- Нейрональные цероидные липофусцинозы

Содержание
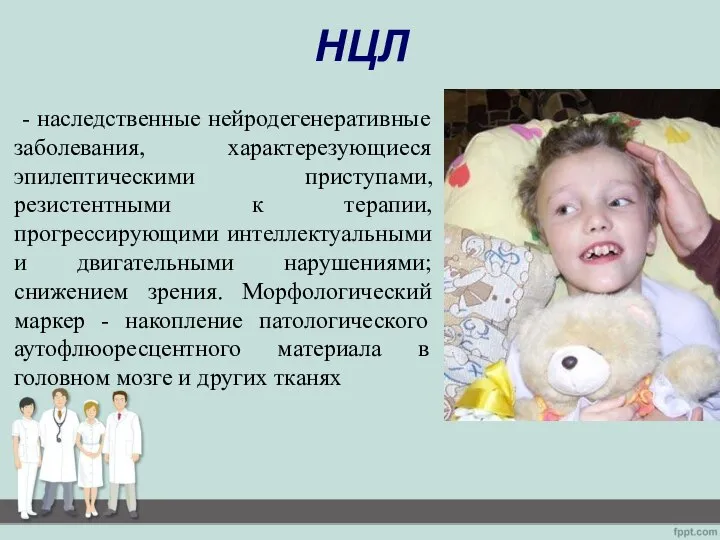
- 2. НЦЛ - наследственные нейродегенеративные заболевания, характерезующиеся эпилептическими приступами, резистентными к терапии, прогрессирующими интеллектуальными и двигательными нарушениями;
- 3. Классификация Формы Врожденная (CNCL) Инфантильная (INCL) Поздняя инфантильная (LINCL) Ювенильная (JNCL) Взрослая(ANCL) Северная эпилепсия
- 4. Типы НЦЛ Также принято разделять НЦЛ на типы в зависимости от первичного молекулярно-генетического дефекта – соответственно
- 5. Историческая справка Первые случаи НЦЛ были описаны еще в начале 19 века. В 1826 г. Кристиан
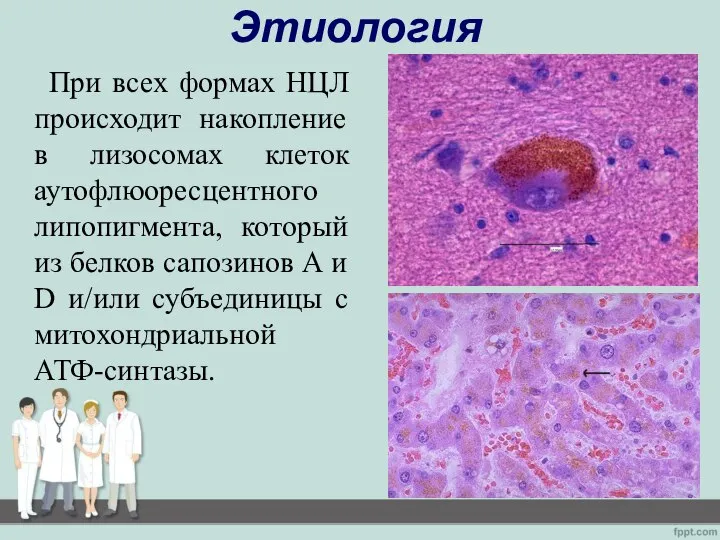
- 6. Этиология При всех формах НЦЛ происходит накопление в лизосомах клеток аутофлюоресцентного липопигмента, который из белков сапозинов
- 7. НЦЛ широко распространены во всем мире и, возможно, являются наиболее частыми заболеваниями из группы наследственных нейродегенеративных
- 8. Врожденный НЦЛ ( НЦЛ 10 тип; CNLC) Впервые была описана Норманом и Вудом в 1941 году.
- 9. Инфантильная форма (НЦЛ1 типа, болезнь Сантавуори-Халтиа, INCL) Была впервые описана Santavuori и соавт. в 1973 г.
- 10. ЭЭГ - уменьшение амплитуды и замедление основного ритма; во время сна – отсутствие сонных веретен. Характерно
- 11. Поздняя инфантильная форма (НЦЛ 2,5,6,7 типов, LINCL) - генетически гетерогенная группа заболеваний, характеризующаяся сходным возрастом начала.
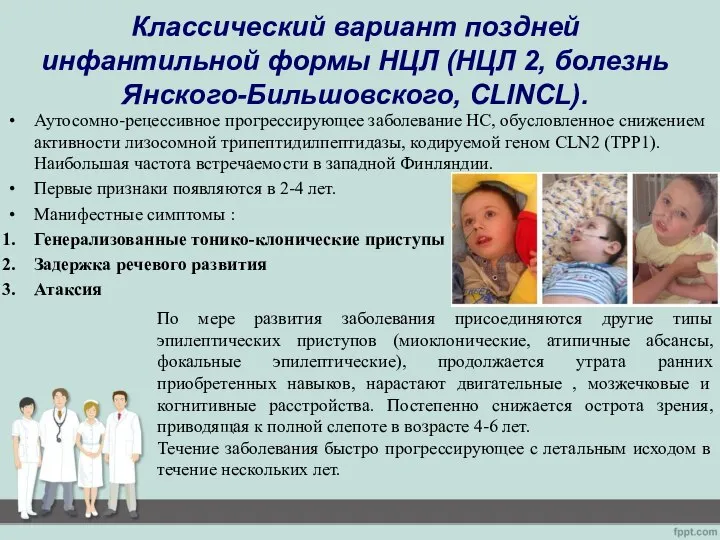
- 12. Классический вариант поздней инфантильной формы НЦЛ (НЦЛ 2, болезнь Янского-Бильшовского, CLINCL). Аутосомно-рецессивное прогрессирующее заболевание НС, обусловленное
- 13. Нейровизуализация – атрофия полушарий головного мозга и мозжечка, степень которой зависит от стадии заболевания. В некоторых
- 14. Ювенильные формы (НЦЛ 3, 9 типов, GNCL) Большая часть случаев ювенильных форм болезни обусловлено мутациями в
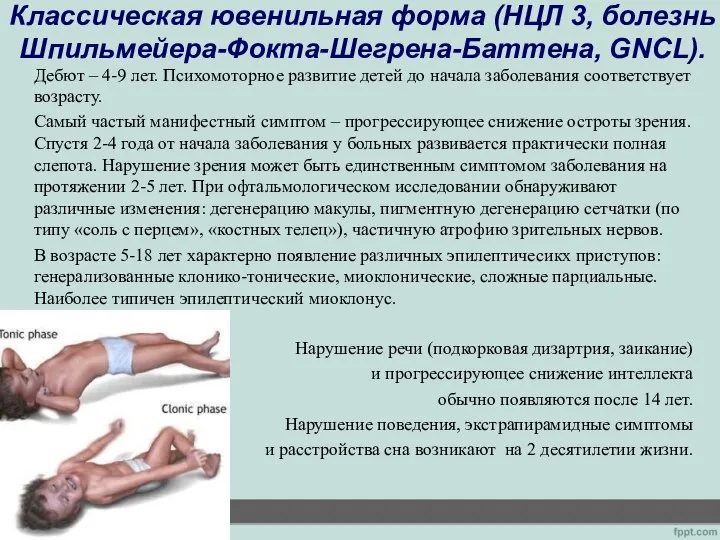
- 15. Классическая ювенильная форма (НЦЛ 3, болезнь Шпильмейера-Фокта-Шегрена-Баттена, GNCL). Дебют – 4-9 лет. Психомоторное развитие детей до
- 16. У некоторых пациентов ведущими в клинике являются когнитивные нарушения и расстройства поведения: нарушение мышления, рассеянное внимание,
- 17. ЭЭГ – дезорганизация корковой ритмики, высокоамплитудная спайковая активность и разряды диффузных медленных волн. Возможно появление длительных
- 18. Взрослая форма НЦЛ (Болезнь Куфса, ANCL) Начальные симптомы в возрасте 30 лет, с быстрым прогрессированием заболевания
- 19. Северная эпилепсия (НЦЛ 8 типа, NORTHERN EPILEPSI, PROGRESSIVE EPILEPSI VIS MENTAL RETARDATOIN (PEMR)) Эта форма НЦЛ
- 20. Верификация диагноза НЦЛ характеризуется возраст- зависимым дебютом, эпилептическими приступами, прогрессирующими двигательными и интеллектуально-мнестическими нарушениями; расстройствами зрения.
- 21. Лечение В настоящее время не разработано эффективное лечение НЦЛ. Применяется симптоматическая терапия. Подбор АЭП начинают с
- 22. В настоящее время продолжаются клинические испытания по применению генотерапии, стволовых клеток, шаперонотерапии и препарата CystagonTM. Все
- 24. Скачать презентацию
Слайд 2НЦЛ
- наследственные нейродегенеративные заболевания, характерезующиеся эпилептическими приступами, резистентными к терапии, прогрессирующими
НЦЛ
- наследственные нейродегенеративные заболевания, характерезующиеся эпилептическими приступами, резистентными к терапии, прогрессирующими
Слайд 3Классификация
Формы
Врожденная (CNCL)
Инфантильная (INCL)
Поздняя инфантильная (LINCL)
Ювенильная (JNCL)
Взрослая(ANCL)
Северная эпилепсия
Классификация
Формы
Врожденная (CNCL)
Инфантильная (INCL)
Поздняя инфантильная (LINCL)
Ювенильная (JNCL)
Взрослая(ANCL)
Северная эпилепсия
Слайд 4Типы НЦЛ
Также принято разделять НЦЛ на типы в зависимости от первичного
Типы НЦЛ
Также принято разделять НЦЛ на типы в зависимости от первичного

Слайд 5Историческая справка
Первые случаи НЦЛ были описаны еще в начале 19 века.
Историческая справка
Первые случаи НЦЛ были описаны еще в начале 19 века.

Слайд 6Этиология
При всех формах НЦЛ происходит накопление в лизосомах клеток аутофлюоресцентного липопигмента,
Этиология
При всех формах НЦЛ происходит накопление в лизосомах клеток аутофлюоресцентного липопигмента,
Слайд 7НЦЛ широко распространены во всем мире и, возможно, являются наиболее частыми заболеваниями
НЦЛ широко распространены во всем мире и, возможно, являются наиболее частыми заболеваниями

Слайд 8Врожденный НЦЛ
( НЦЛ 10 тип; CNLC)
Впервые была описана Норманом и Вудом
Врожденный НЦЛ
( НЦЛ 10 тип; CNLC)
Впервые была описана Норманом и Вудом

Слайд 9Инфантильная форма
(НЦЛ1 типа, болезнь Сантавуори-Халтиа, INCL)
Была впервые описана Santavuori и соавт.
Инфантильная форма
(НЦЛ1 типа, болезнь Сантавуори-Халтиа, INCL)
Была впервые описана Santavuori и соавт.

Слайд 10 ЭЭГ - уменьшение амплитуды и замедление основного ритма; во время сна
ЭЭГ - уменьшение амплитуды и замедление основного ритма; во время сна

Слайд 11Поздняя инфантильная форма (НЦЛ 2,5,6,7 типов, LINCL)
- генетически гетерогенная группа заболеваний,
Поздняя инфантильная форма (НЦЛ 2,5,6,7 типов, LINCL)
- генетически гетерогенная группа заболеваний,

Слайд 12Классический вариант поздней инфантильной формы НЦЛ (НЦЛ 2, болезнь Янского-Бильшовского, CLINCL).
Аутосомно-рецессивное прогрессирующее
Классический вариант поздней инфантильной формы НЦЛ (НЦЛ 2, болезнь Янского-Бильшовского, CLINCL).
Аутосомно-рецессивное прогрессирующее
Слайд 13Нейровизуализация – атрофия полушарий головного мозга и мозжечка, степень которой зависит от
Нейровизуализация – атрофия полушарий головного мозга и мозжечка, степень которой зависит от

Слайд 14Ювенильные формы
(НЦЛ 3, 9 типов, GNCL)
Большая часть случаев ювенильных форм
Ювенильные формы
(НЦЛ 3, 9 типов, GNCL)
Большая часть случаев ювенильных форм

Слайд 15Классическая ювенильная форма (НЦЛ 3, болезнь Шпильмейера-Фокта-Шегрена-Баттена, GNCL).
Дебют – 4-9 лет.
Классическая ювенильная форма (НЦЛ 3, болезнь Шпильмейера-Фокта-Шегрена-Баттена, GNCL).
Дебют – 4-9 лет.
Слайд 16 У некоторых пациентов ведущими в клинике являются когнитивные нарушения и расстройства
У некоторых пациентов ведущими в клинике являются когнитивные нарушения и расстройства

Слайд 17ЭЭГ – дезорганизация корковой ритмики, высокоамплитудная спайковая активность и разряды диффузных медленных
ЭЭГ – дезорганизация корковой ритмики, высокоамплитудная спайковая активность и разряды диффузных медленных

Слайд 18Взрослая форма НЦЛ
(Болезнь Куфса, ANCL)
Начальные симптомы в возрасте 30 лет,
Взрослая форма НЦЛ
(Болезнь Куфса, ANCL)
Начальные симптомы в возрасте 30 лет,

Слайд 19Северная эпилепсия (НЦЛ 8 типа, NORTHERN EPILEPSI, PROGRESSIVE EPILEPSI VIS MENTAL RETARDATOIN
Северная эпилепсия (НЦЛ 8 типа, NORTHERN EPILEPSI, PROGRESSIVE EPILEPSI VIS MENTAL RETARDATOIN

Слайд 20Верификация диагноза
НЦЛ характеризуется возраст- зависимым дебютом, эпилептическими приступами, прогрессирующими двигательными и
Верификация диагноза
НЦЛ характеризуется возраст- зависимым дебютом, эпилептическими приступами, прогрессирующими двигательными и

Слайд 21Лечение
В настоящее время не разработано эффективное лечение НЦЛ.
Применяется симптоматическая терапия.
Подбор
Лечение
В настоящее время не разработано эффективное лечение НЦЛ.
Применяется симптоматическая терапия.
Подбор

Слайд 22В настоящее время продолжаются клинические испытания по применению генотерапии, стволовых клеток, шаперонотерапии
В настоящее время продолжаются клинические испытания по применению генотерапии, стволовых клеток, шаперонотерапии

 Қалқанша безінің,ұйқы безінің,бүйрек үсті безінің гормондары
Қалқанша безінің,ұйқы безінің,бүйрек үсті безінің гормондары Николай Иванович Пирогов. Русский хирург и анатом
Николай Иванович Пирогов. Русский хирург и анатом ПМП при клинической смерти. Основные инфекционные заболевания, их классификация и профилактика
ПМП при клинической смерти. Основные инфекционные заболевания, их классификация и профилактика Pathologic and non pathologic germs in human organism
Pathologic and non pathologic germs in human organism Неотложные состояния 2
Неотложные состояния 2 Ерте және жасырын түрдегі ауруларды
Ерте және жасырын түрдегі ауруларды Кровотечение и его виды
Кровотечение и его виды Туберкулез. Збудник
Туберкулез. Збудник La salute
La salute Функции крови
Функции крови Осложнение инъекций
Осложнение инъекций Цифровая кардиология
Цифровая кардиология External fixation devices. Volkov-Hovhannisyan Apparatus
External fixation devices. Volkov-Hovhannisyan Apparatus Цереброваскулярные заболевания
Цереброваскулярные заболевания Обгрунтування складу фармацевтичної композиції для лікування нейродегенеративних захворювань
Обгрунтування складу фармацевтичної композиції для лікування нейродегенеративних захворювань Лекарственные средства, влияющие на функцию матки
Лекарственные средства, влияющие на функцию матки Тонкощі використання інсектоакариців у дрібних тварин
Тонкощі використання інсектоакариців у дрібних тварин БА -– это хроническое воспалительное заболевание дыхательных путей
БА -– это хроническое воспалительное заболевание дыхательных путей Доврачебная помощь при аллергических реакциях и анафилактическом шоке
Доврачебная помощь при аллергических реакциях и анафилактическом шоке Профессия патологоанатома
Профессия патологоанатома Гимнастика для глаз
Гимнастика для глаз Шок. Кардиогенный шок
Шок. Кардиогенный шок Классификация нарушений спинномозгового кровообращения. Диагностика и дифференциальная диагностика
Классификация нарушений спинномозгового кровообращения. Диагностика и дифференциальная диагностика Аритмии и блокады
Аритмии и блокады Ревматоидный артрит
Ревматоидный артрит Нейссерии. Род Neisseria
Нейссерии. Род Neisseria Волевая ликвидация глубокого дыхания. Метод ВЛГД
Волевая ликвидация глубокого дыхания. Метод ВЛГД Лейкоз (белокровие)
Лейкоз (белокровие)